liuyujie0136's Website
A website for self learning, collecting and sharing.
使用deeptools处理BAM文件
deepTools Docs
https://www.jianshu.com/p/b494426ecd32
deeptools是基于Python开发的一套工具,用于处理诸如RNA-seq, ChIP-seq, MNase-seq, ATAC-seq等高通量数据。工具分为四个模块:
- BAM和bigWig文件处理
- 质量控制
- 热图和其他描述性作图
- 其他
当然也可以简单分为两个部分:数据处理和可视化。
对于deeptools里的任意子命令,都支持--help看帮助文档,--numberOfProcessors/-p设置多核处理,--region/-r CHR:START:END处理部分区域。还有一些过滤用参数部分子命令可用,如ignoreDuplicates, minMappingQuality, samFlagInclude, samFlagExclue.

BAM转换为bigWig或bedGraph
BAM文件是SAM的二进制转换版,应该都知道。那么bigWig格式是什么?bigWig是wig或bedGraph的二进制版,存放区间的坐标轴信息和相关计分(score),主要用于在基因组浏览器上查看数据的连续密度图,可用wigToBigWig从wiggle进行转换。
bedGraph和wig格式是什么? USCS的帮助文档称这两个格式数是已经过时的基因组浏览器图形轨展示格式,前者展示稀松型数据,后者展示连续性数据。目前推荐使用bigBed/bigWig这两种格式取代前两者。
为什么要用bigWig? 主要是因为BAM文件比较大,直接用于展示时对服务器要求较大。因此在GEO上仅会提供bw,即bigWig下载,便于下载和查看。如果真的感兴趣,则可以下载原始数据进行后续分析。
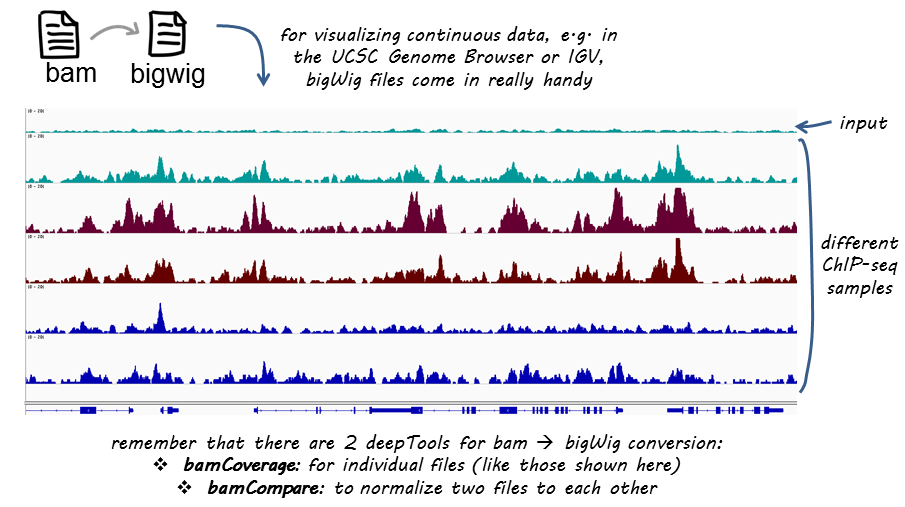
deeptools提供bamCoverage和bamCompare进行格式转换,为了能够比较不同的样本,需要对先将基因组分成等宽分箱(bin),统计每个分箱的read数,最后得到描述性统计值。对于两个样本,描述性统计值可以是两个样本的比率,或是比率的log2值,或者是差值。如果是单个样本,可以用SES方法进行标准化。
bamCoverage的基本用法
bamCoverage \
-e 170 \ # --extendReads
-bs 10 \ # --binSize
-b exp_rep1_1.sorted.bam \ # --bam
-o exp_rep1_1.bw # --outFileName
得到的bw文件就可以送去IGV/Jbrowse进行可视化。 这里的参数仅使用了-e/--extendReads和-bs/--binSize即拓展了原来的read长度,且设置分箱的大小。其他参数还有
--filterRNAstrand {forward, reverse}: 仅统计指定正链或负链--region/-r CHR:START:END: 选取某个区域统计--smoothLength: 通过使用分箱附近的read对分箱进行平滑化
如果为了其他结果进行比较,还需要进行标准化,deeptools提供了如下参数:
--scaleFactor: 缩放系数--effectiveGenomeSize: 有效染色体大小,用于RPGC标准化--normalizeUsing {RPKM, CPM, BPM, RPGC, None}: 标准化--ignoreForNormalization: 指定那些染色体不需要经过标准化
如果需要以100为分箱,并且用RPKM标准化,且仅统计某一条染色体区域的正链,输出格式为bedgraph,那么命令行可以这样写
bamCoverage \
-e 170 \
-bs 100 \
-of bedgraph \ # --outFileFormat
-r Chr4:12985884:12997458 \ # --region
--filterRNAstrand forward \
--normalizeUsing RPKM \
-b exp_rep1_1.sorted.bam \
-o exp_rep1_1.bedGraph
bamCompare和bamCoverage类似,只不过需要提供两个样本,并且采用SES方法进行标准化,于是多了--ratio参数。
多样本分析
这部分内容主要分析处理组不同重复间的相关程度,会用到multiBamSummary、plotCorrelation和plotPCA三个模块。主要目的是看下对照组和处理组中的组间差异和组内相似性。
# 如果已经把BAM转换成BW, 可以直接用multiBigWigSummary
# 统计reads在全基因组范围的情况
multiBamSummary bins \
-e 130 \
-bs 1000 \
-o treat_results.npz \
--bamfiles \
exp_rep1_1.sorted.bam \
exp_rep1_2.sorted.bam \
exp_rep2_1.sorted.bam \
exp_rep2_2.sorted.bam \
ctrl_rep1_1.sorted.bam \
ctrl_rep1_2.sorted.bam \
ctrl_rep2_1.sorted.bam \
ctrl_rep2_2.sorted.bam
# 散点图
plotCorrelation \
-in treat_results.npz \
-o treat_results.png \
--corMethod spearman \
-p scatterplot
# 热图
plotCorrelation \
-in treat_results.npz \
-o treat_results_heatmap.png \
--corMethod spearman \
-p heatmap
# 主成分分析
plotPCA \
-in treat_results.npz \
-o pca.png
peak分布可视化
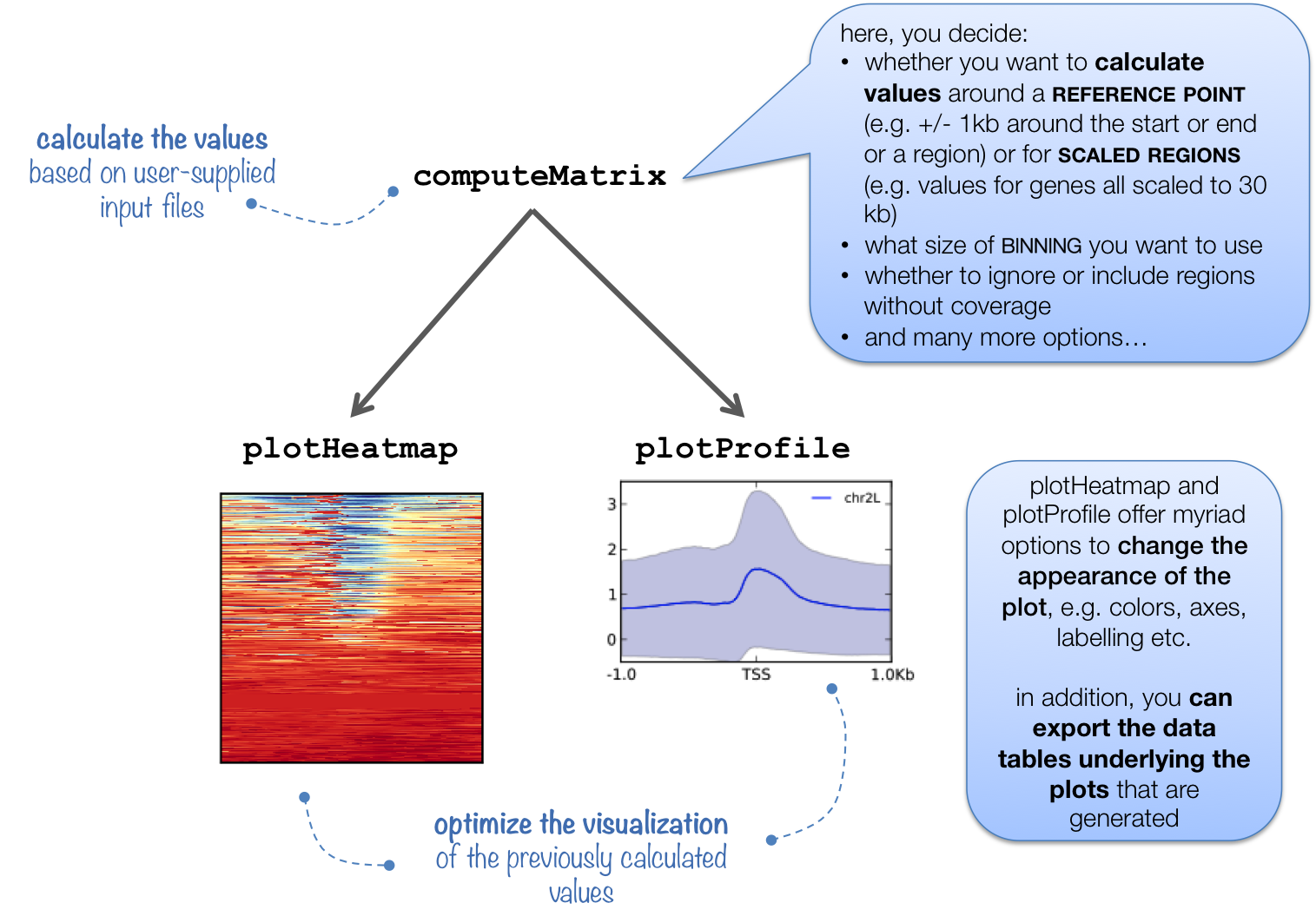
为了统计全基因组范围的peak在基因特征的分布情况,需要用到computeMatrix计算,用plotHeatmap以热图的方式对覆盖进行可视化,用plotProfile以折线图的方式展示覆盖情况。
computeMatrix具有两个模式:scale-region和reference-point。前者用来信号在一个区域内分布,后者查看信号相对于某一个点的分布情况。
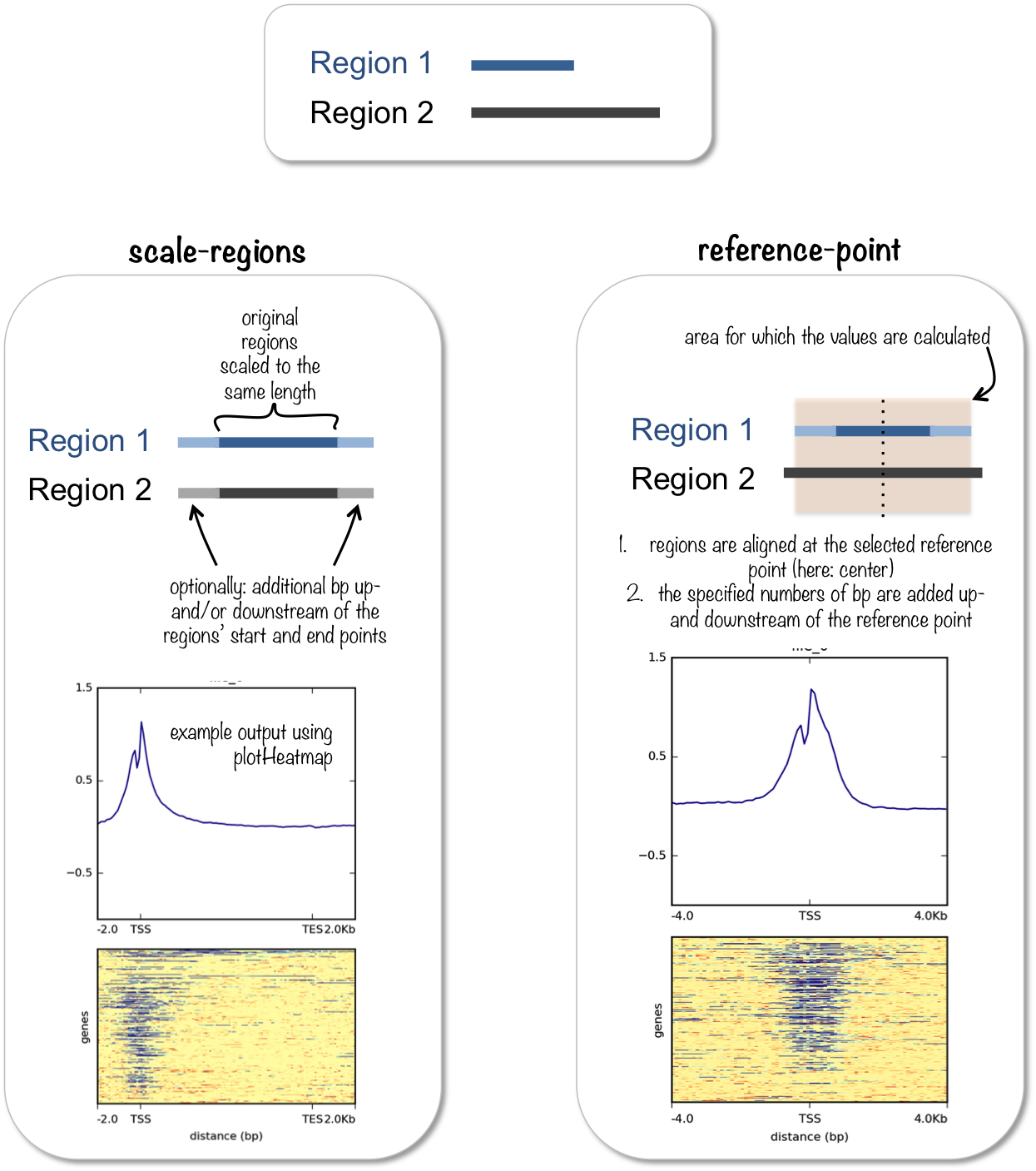
无论是那个模式,都有有两个参数是必须的,-S是提供bigwig文件,-R是提供基因的注释信息。
scale-regions模式
computeMatrix scale-regions \
-b 3000 -a 5000 \ # 感兴趣的区域,-b上游,-a下游
-R TAIR10_GFF3_genes.bed \
-S exp_rep1_1.bw \
--skipZeros \
--o exp_rep1_1.genes.matrix.gz\
--outFileNameMatrix exp_rep1_1.genes.tab \
--outFileSortedRegions exp_rep1_1.genes.bed
reference-point模式
computeMatrix reference-point \
--referencePoint TSS \ # 选择参考点: TES, center
-b 3000 -a 5000 \
-R TAIR10_GFF3_genes.bed \
-S exp_rep1_1.bw \
--skipZeros \
-o exp_rep1_1.TSS.matrix.gz
结果可视化
可视化的方法有两种,一种是轮廓图,一种是热图。两则都提供了足够多的参数对结果进行细节上的修改。
plotProfile \
-m exp_rep1_1.TSS.matrix.gz \
-o ExampleProfile.png \
--numPlotsPerRow 2 \
--plotTitle "ExampleProfile"
plotHeatmap \
-m exp_rep1_1.TSS.matrix.gz \
-o ExampleHeatmap.png \
示例代码
file=$1;
pre=`basename $file |cut -d '.' -f 1`;
# small RNA
computeMatrix reference-point \
--referencePoint center \
-b 200 -a 200 \
--bs 10 \
-R $file \
-S WT_sRNA.bw Mutant_sRNA.bw \
-p 8 \
-o $pre.sRNA.matrix.gz
plotProfile \
-m $pre.sRNA.matrix.gz \
-o $pre.sRNA.pdf \
--perGroup
# CHH methylation
computeMatrix reference-point \
--referencePoint TSS \
-b 2000 -a 4000 \
--bs 100 \
-R $file \
-S WT_CHH.bw Mutant_CHH.bw \
-p 8 \
--nanAfterEnd \
-o $pre.CHH.TSS.matrix.gz
plotProfile \
-m $pre.TSS.CHH.matrix.gz \
-o $pre.TSS.CHH.pdf \
--perGroup