liuyujie0136's Website
A website for self learning, collecting and sharing.
学习一遍ChIPseeker的使用
https://www.jianshu.com/p/c76e83e6fa57
刘小泽写于2020.5.23-24
Y叔的原文在:https://mp.weixin.qq.com/s/3CMj0xejiV-FSMC-Vxd_-w
0 ChIPseeker的诞生
Y叔一开始使用ChIPpeakAnno进行注释,但使用UCSC genome browser检验结果的时候,发现对不上;另外之前在使用ChIPpeakAnno过程中写了一些可视化函数。后来经过漫长的半夜宿舍苦战,写出了ChIPseeker
1 ChIP-seq简介
ChIP是指染色质免疫沉淀,它通特异结合抗体将DNA结合蛋白免疫沉淀,可以用于捕获蛋白质(如转录因子,组蛋白修饰)的DNA靶点。之前结合芯片就有ChIP-on-chip,后来二代测序加持诞生了ChIP-seq。优点是:不再需要设计探针(探针往往存在着一定的偏向性)
2007年来自三个不同的实验室,几乎是同时间出来(最长差不了3个月),分别发CNS,一起定义了这个ChIPseq技术
- Johnson DS, Mortazavi A et al. (2007) Genome-wide mapping of in vivo protein–DNA interactions. Science 316: 1497–1502
- Robertson G et al.(2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods 4: 651–657
- Schmid et al. (2007) ChIP-Seq Data reveal nucleosome architecture of human promoters. Cell 131: 831–832
主要有4步:Cross-linking、Sonication、IP、Sequencing
简而言之是:DNA和蛋白质交联(cross-linking)、超声(sonication)将染色体随机切割、利用抗原抗体的特异性识别(IP)、把目标蛋白相结合的DNA片段沉淀下来,反交联释放DNA片段,最后是测序(sequencing)
分析流程示例图1:
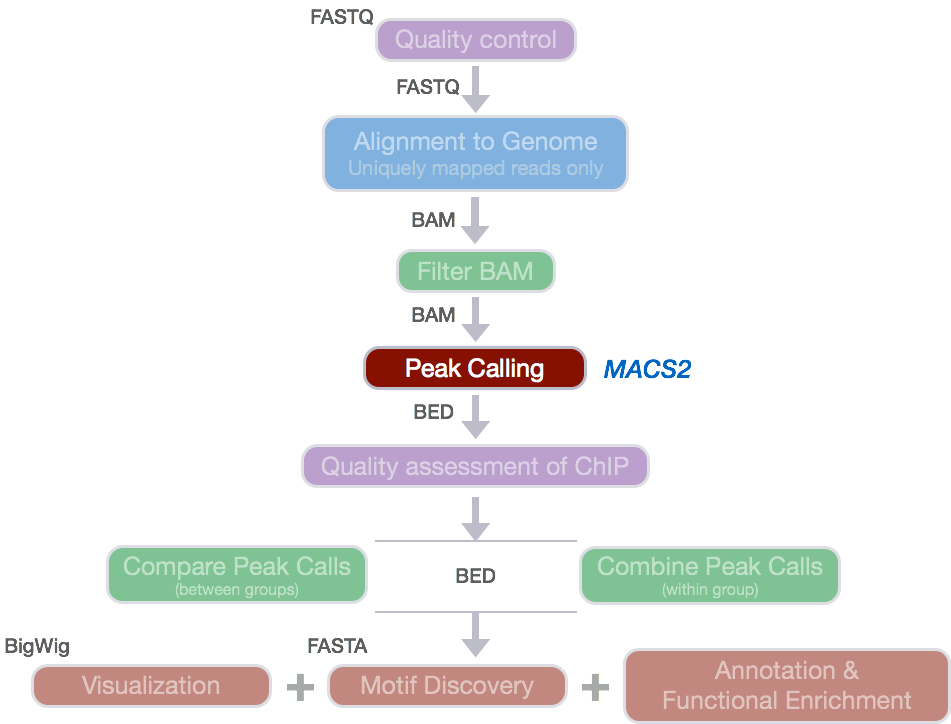
分析流程示例图2:
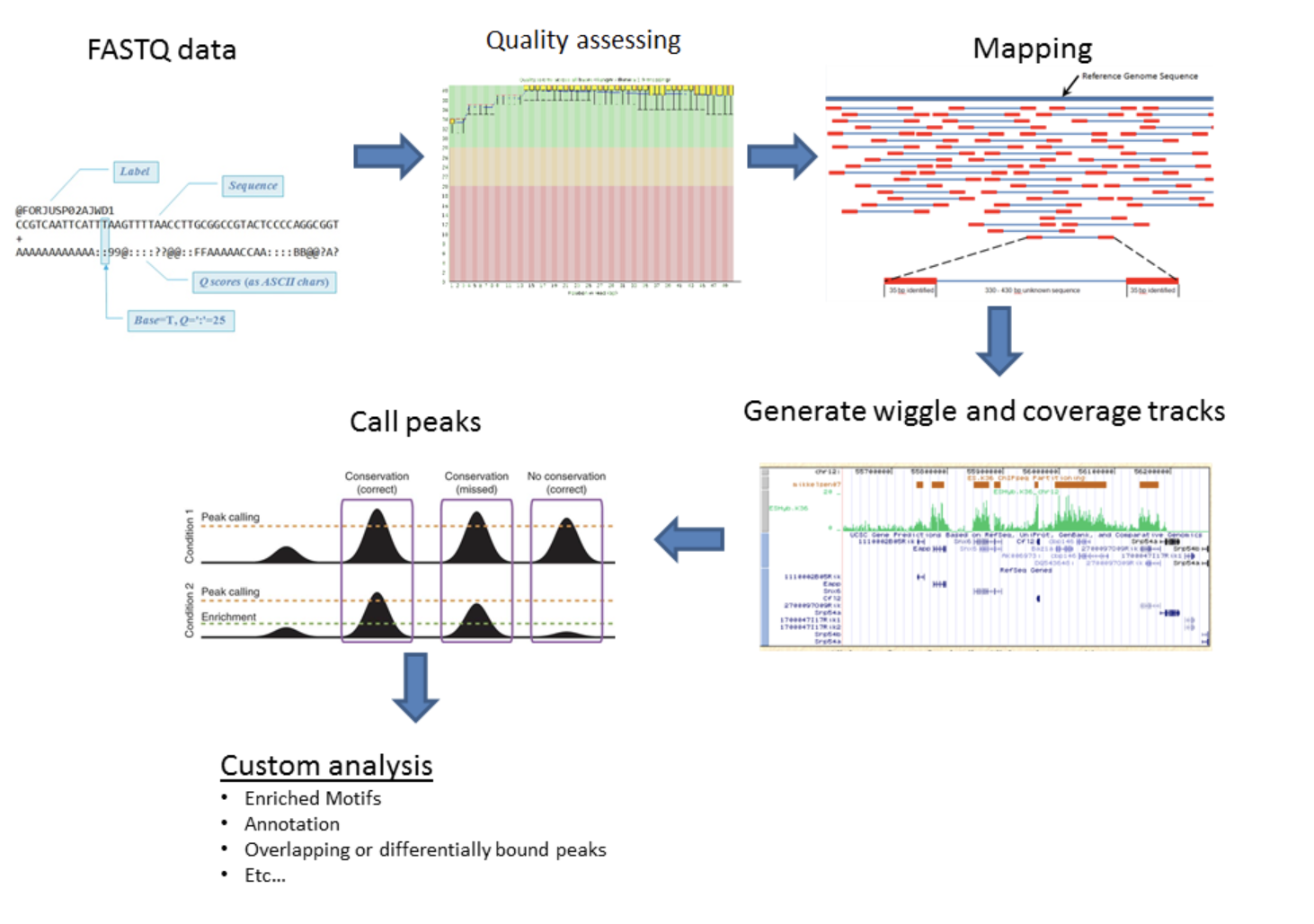
原始数据=》质控=》比对=》拿到DNA片段在染色体上的位置信息=》peak calling (去除背景噪音)=》拿到peaks(protein binding site)=》下游分析(可视化、找相关基因、motif分析等等)
2 必须知晓的BED文件
全称是:Browser Extensible Data,为基因组浏览器而生
包括3个必须字段和9个可选字段:
3个必须
- 1 chrom - 染色体名字
- 2 chromStart - 染色体起始位点(起始于0,而不是1)许多软件忽略了这一点,存在一个碱基的位移(如peakAnalyzer, ChIPpeakAnno存在这个问题),Homer、ChIPseeker没有
- 3 chromEnd - 染色体终止位点
9个可选
- 4 name - 名字
- 5 score - 分值(0-1000), 用于genome browser展示时上色。
- 6 strand - 正负链,对于ChIPseq数据来说,一般没有正负链信息。
- 7 thickStart - 画矩形的起点
- 8 thickEnd - 画矩形的终点
- 9 itemRgb - RGB值
- 10 blockCount - 子元件(比如外显子)的数目
- 11 blockSizes - 子元件的大小
- 12 blockStarts - 子元件的起始位点
一般只用前5个足矣(MACS的输出结果也是前5个字段)
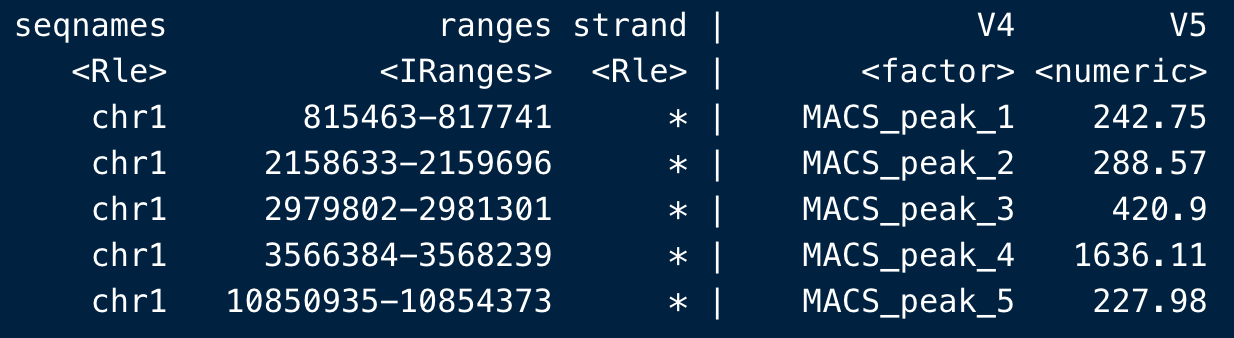
第5列score的含义是:the summit height of fragment pileup. 也即是片段堆积的峰高
3 使用covplot可视化BED数据
一般拿到数据后,会先可视化一下数据的全景
# 自带示例数据(这也是Bioconductor包的一个特点,提交R包需要有说明书和测试数据)
library(ChIPseeker)
library(ggplot2)
files <- getSampleFiles()
# 有5个文件
> basename(unlist(files))
[1] "GSM1174480_ARmo_0M_peaks.bed.gz"
[2] "GSM1174481_ARmo_1nM_peaks.bed.gz"
[3] "GSM1174482_ARmo_100nM_peaks.bed.gz"
[4] "GSM1295076_CBX6_BF_ChipSeq_mergedReps_peaks.bed.gz"
[5] "GSM1295077_CBX7_BF_ChipSeq_mergedReps_peaks.bed.gz"
covplot(files[[5]])
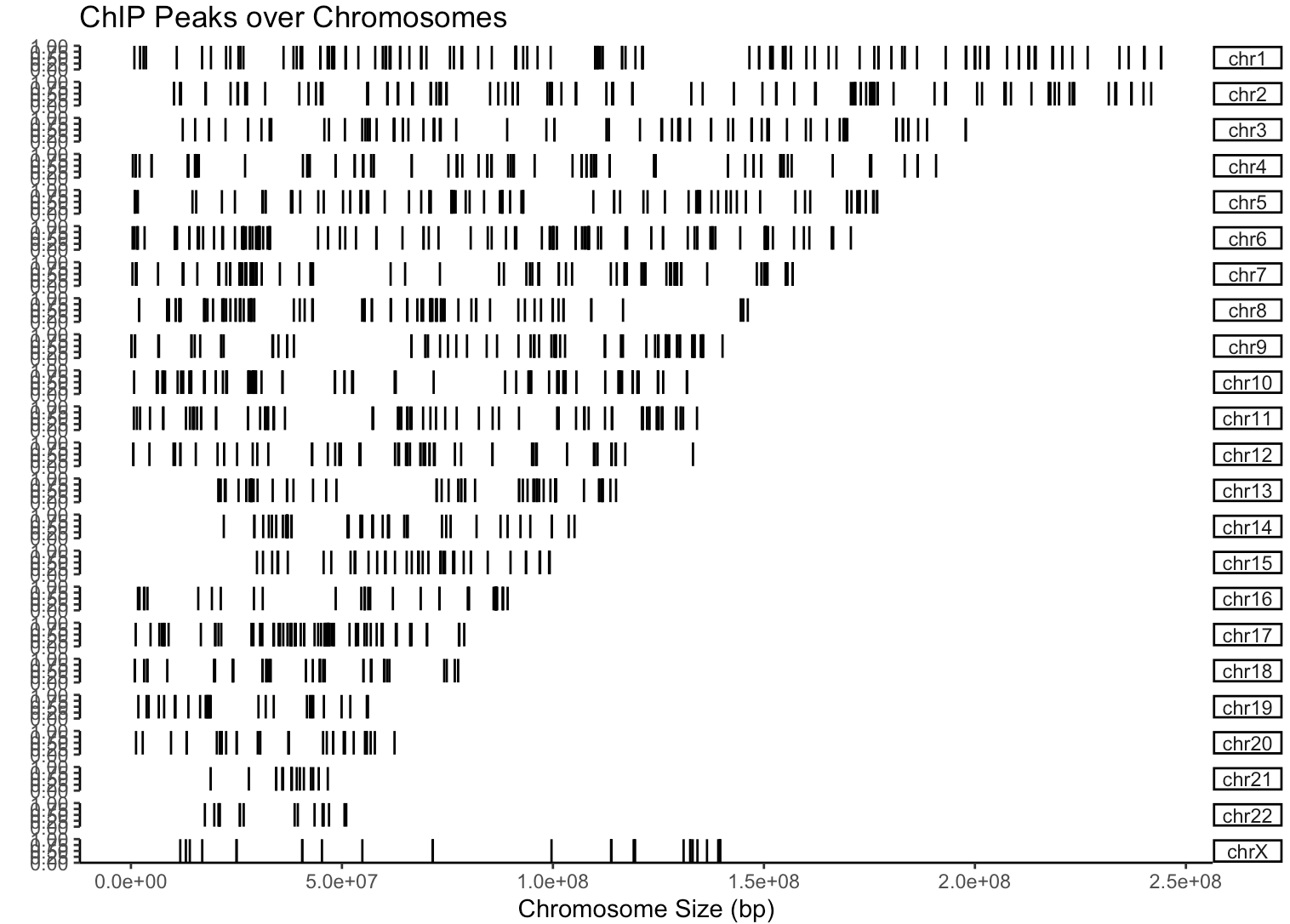
还支持多个文件同时画
只要转为GRanges对象即可
# 比如要画第4、5个文件(MACS生成的BED文件包含常规的5列)
peak=GenomicRanges::GRangesList(CBX6=readPeakFile(files[[4]]),CBX7=readPeakFile(files[[5]]))
画图
covplot(peak, weightCol="V5") + facet_grid(chr ~ .id)
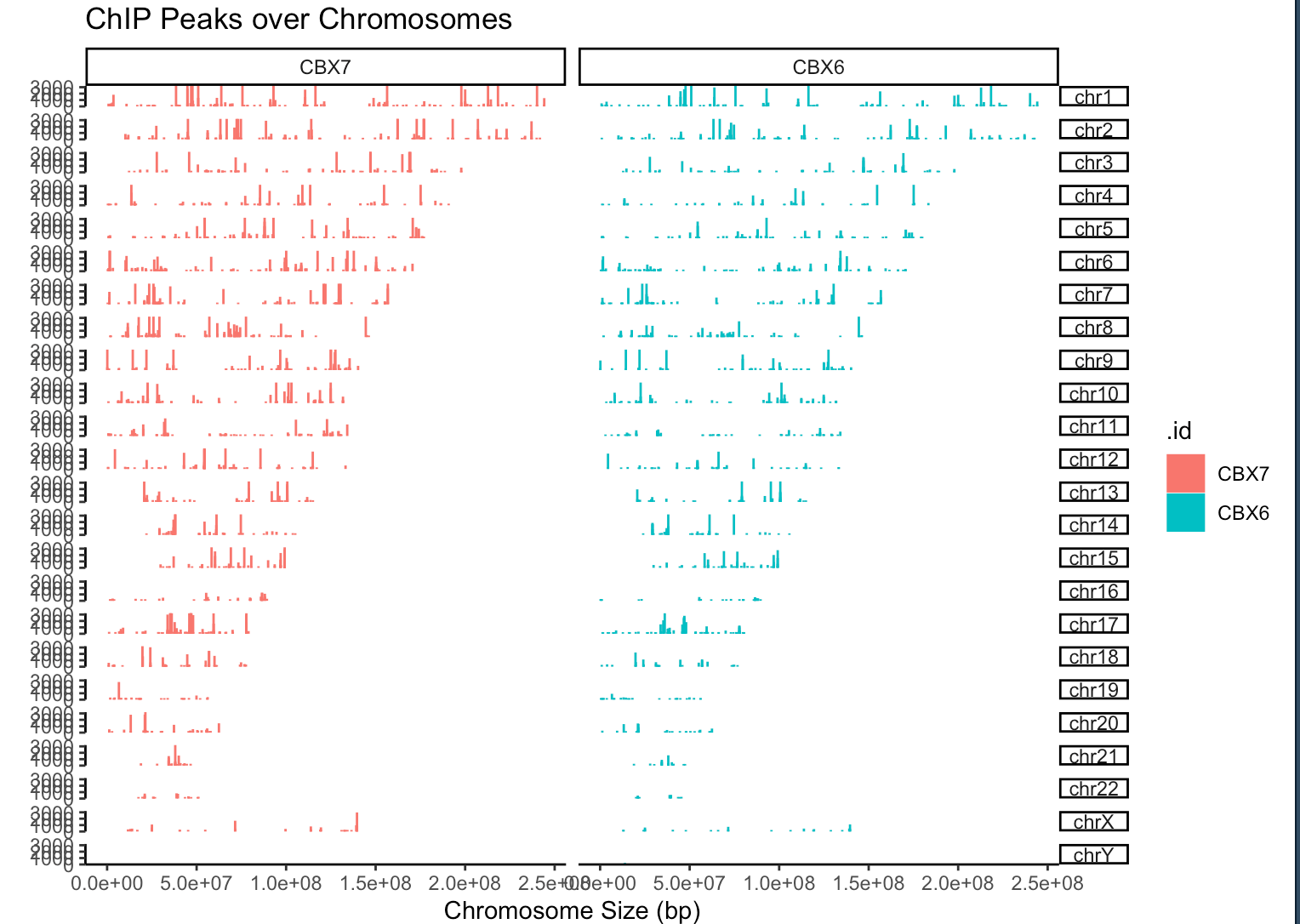
取小区间,例如只取几条染色体,还能定义染色体的区间大学
covplot(peak, weightCol="V5", chrs=c("chr17", "chr18"),
xlim=c(4e7, 5e7)) + facet_grid(chr ~ .id)
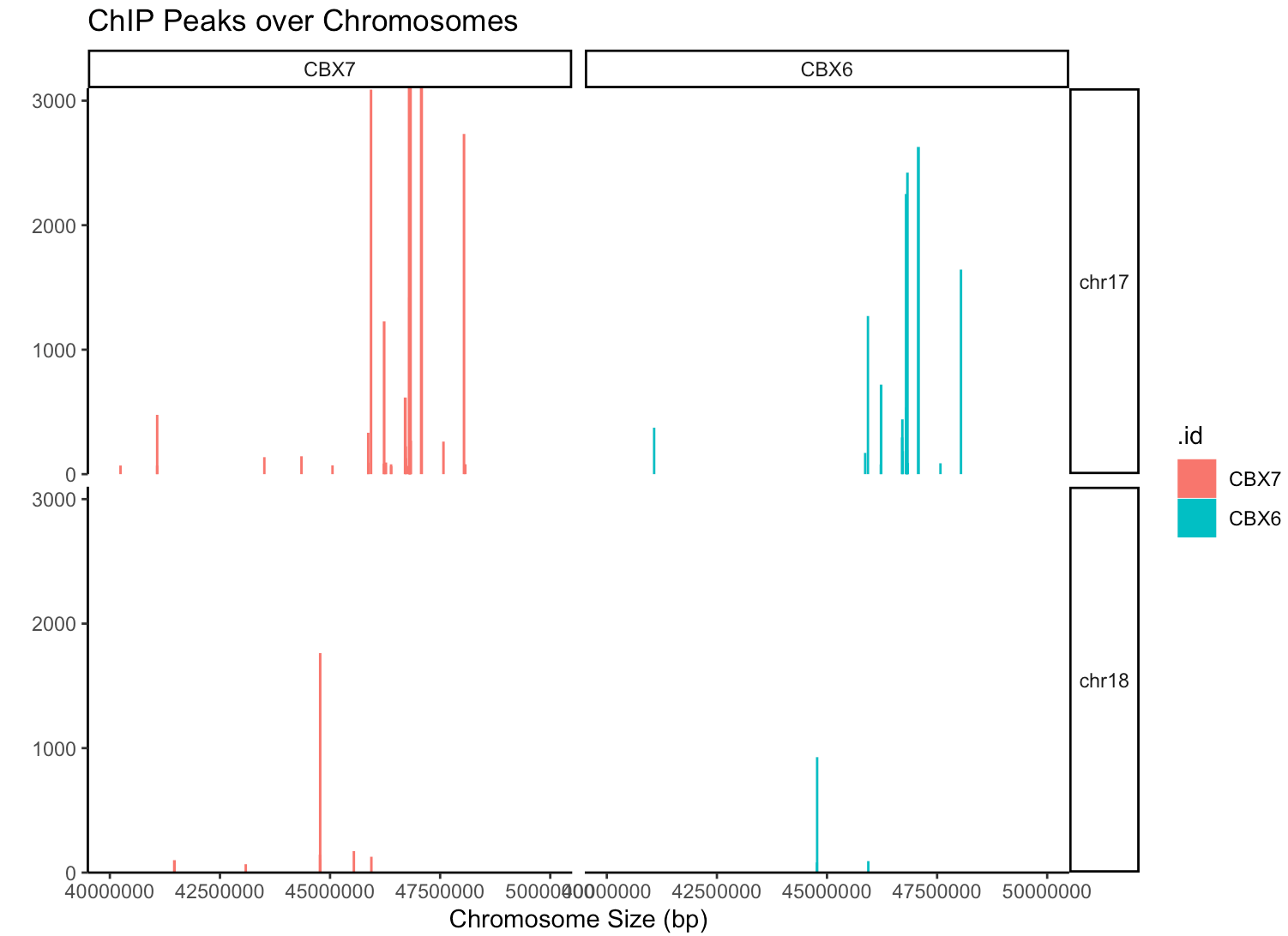
在看完数据全景之后,就会想知道这些peaks和什么类型的基因有关
4 annotatePeak进行peaks的注释
需要使用BED文件(作为query)+注释文件(作为target)
重点是如何获取注释文件
注释信息一般要包含基因的起始终止,基因的外显子、内含子及它们的起始终止、非编码区域位置、功能元件的位置等
ChIPseeker没有物种限制,但前提是物种本身有这些注释信息(不能说物种连参考基因组也没有,那就真的是巧妇难为无米之炊)
需要一个TxDb对象,例如TxDb.Hsapiens.UCSC.hg19.knownGene,然后ChIPseeker就会从中提取信息
# 三步走(提供TxDb注释、提供bed文件、进行注释)
require(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb = TxDb.Hsapiens.UCSC.hg19.knownGene
f = getSampleFiles()[[4]]
x = annotatePeak(f, tssRegion=c(-1000, 1000), TxDb=txdb)
看到这里有个参数tssRegion ,它指定了启动子区域(而启动子区域是没有明确定义的,需要自己指定,这里指定了上下游1kb)
看一下大体结果:
> x
Annotated peaks generated by ChIPseeker
1331/1331 peaks were annotated
Genomic Annotation Summary:
Feature Frequency
9 Promoter 48.1592787
4 5' UTR 0.7513148
3 3' UTR 4.2073629
1 1st Exon 0.7513148
7 Other Exon 3.9068370
2 1st Intron 6.5364388
8 Other Intron 4.8835462
6 Downstream (<=300) 1.1269722
5 Distal Intergenic 29.6769346
看一下详细结果:
as.GRanges(x) %>% head(3)
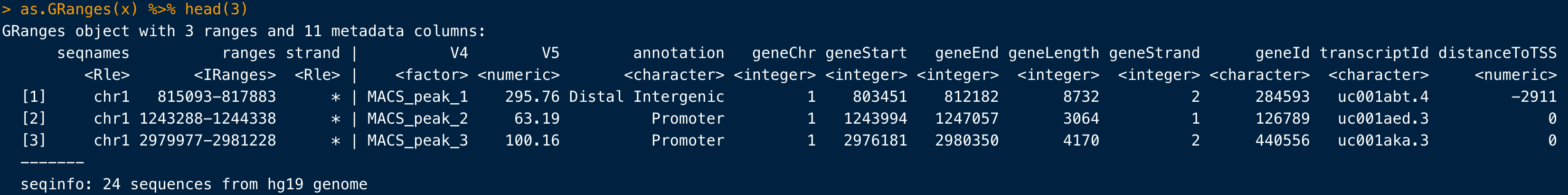
可以转为数据框,方便输出:
tmp=as.data.frame(x)
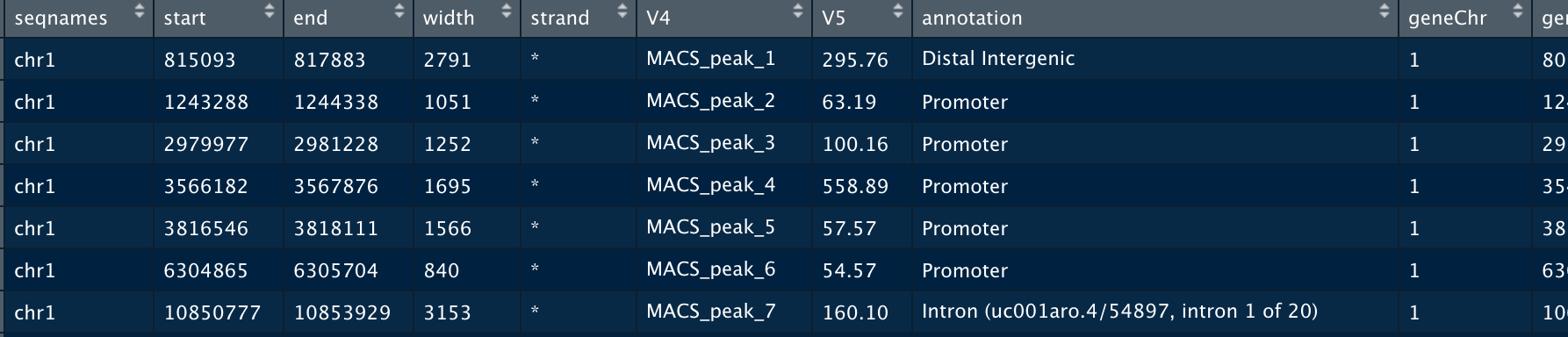
关于注释的类型:
注释类型一:genomic annotation(annotation这一列)
指peak在基因组的位置:落在什么地方,例如外显子、内含子或是UTR
注释类型二:nearest gene annotation(annotation后面的列)
指peak最近的基因:不管peak落在内含子、基因间区还是其他位置,按照peak相对于转录起始位点的距离,都能找到一个离它最近的基因【一般做基因表达调控的,会关注promoter区域,离结合位点最近的基因更可能被调控】
这个距离是根据转录起始位点来计算,一个基因具有多个转录本,因此一个基因可能有多个转录起始位点。注释的结果就会看到有一列是转录本ID
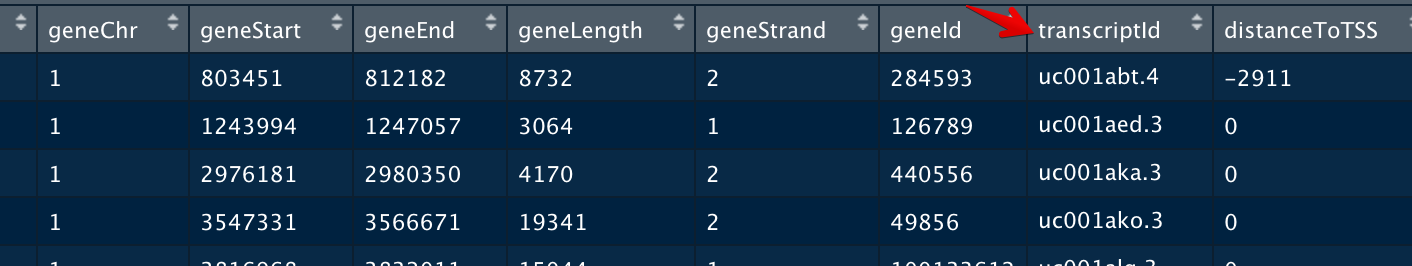
注释类型三:flankDistance(三列: flank_txIds, flank_geneIds和flank_gene_distances)
指peak上下游某个范围内(比如-5kb《=》5kb范围内)都有什么基因
# 传个参数flankDistance
x2 = annotatePeak(f, tssRegion=c(-1000, 1000), TxDb=txdb, addFlankGeneInfo=TRUE, flankDistance=5000)
让基因名变得友好
上面得到的结果都是以geneId(Entrez ID)给出,如果想要Symbol名称,可以再传参数annoDb
library(org.Hs.eg.db)
x3 = annotatePeak(f, tssRegion=c(-1000, 1000), TxDb=txdb,
addFlankGeneInfo=TRUE, flankDistance=5000,
annoDb = "org.Hs.eg.db")
tmp3=as.data.frame(x3)
会再增加3列:ENSEMBL、SYMBOL、GENENAME(如果这里使用的TxDb是Ensemble ID,那么结果就会是Entrez ID、SYMBOL、GENENAME三列)
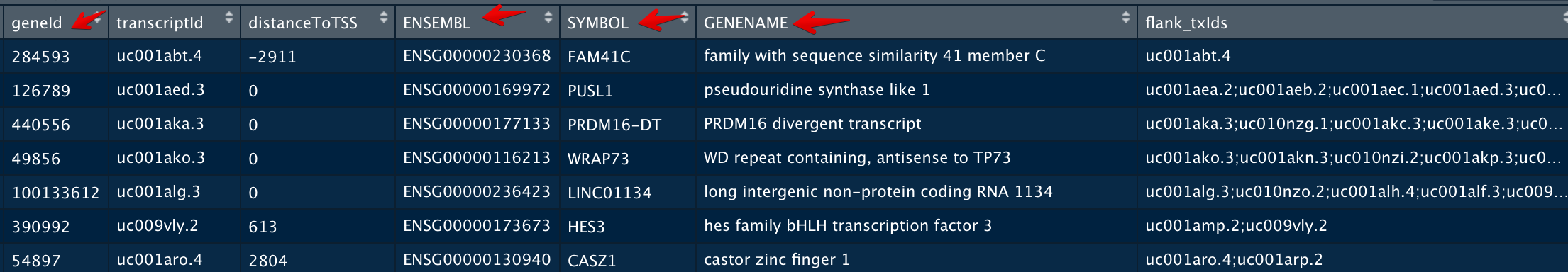
按正负链分开注释
一般ChIPseq数据通常情况下是没有正负链信息的(有特殊的实验可以有)
但如果要做,可以先给peaks分别赋予正负链的信息,然后指定参数sameStrand=TRUE 并分别做两次
这个参数的意思是:(logical)whether find nearest/overlap gene in the same strand
只注释基因的上游或下游
提供了ignoreDownstream和 ignoreUpstream,默认是FALSE
关于TxDb的知识
上面一起操作的前提是物种本身有这些注释信息,而注释信息主要是用TxDb
同一物种的不同版本TxDb
例如TxDb.Hsapiens.UCSC.hg19.knownGene和TxDb.Hsapiens.UCSC.hg38.knownGene 的注释结果是不同的,不能混用。用哪个取决于上游分析比对使用的哪个版本的基因组
不同的版本中基因坐标是不一样的,如果硬要替换,可以使用liftOver将基因组版本坐标进行转换
支持多少物种?
Bioconductor上有30个左右TxDb,也只能覆盖一小部分物种(https://bioconductor.org/packages/3.11/data/annotation/),但UCSC和Ensemble的基因组都可以被ChIPseeker支持,因此所有物种都支持
除了基因注释还能注释lincRNA
require("TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts")
linc_txdb=TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts
x=annotatePeak(peak, TxDb=linc_txdb)
as.GRanges(x)
如何自己制作TxDb?
使用GenomicFeatures包来制作TxDb对象
- makeTxDbFromUCSC: 通过UCSC在线制作TxDb
- makeTxDbFromBiomart: 通过ensembl在线制作TxDb
- makeTxDbFromGRanges:通过GRanges对象制作TxDb
- makeTxDbFromGFF:通过解析GFF文件制作TxDb
比如在线从UCSC生成TxDb:
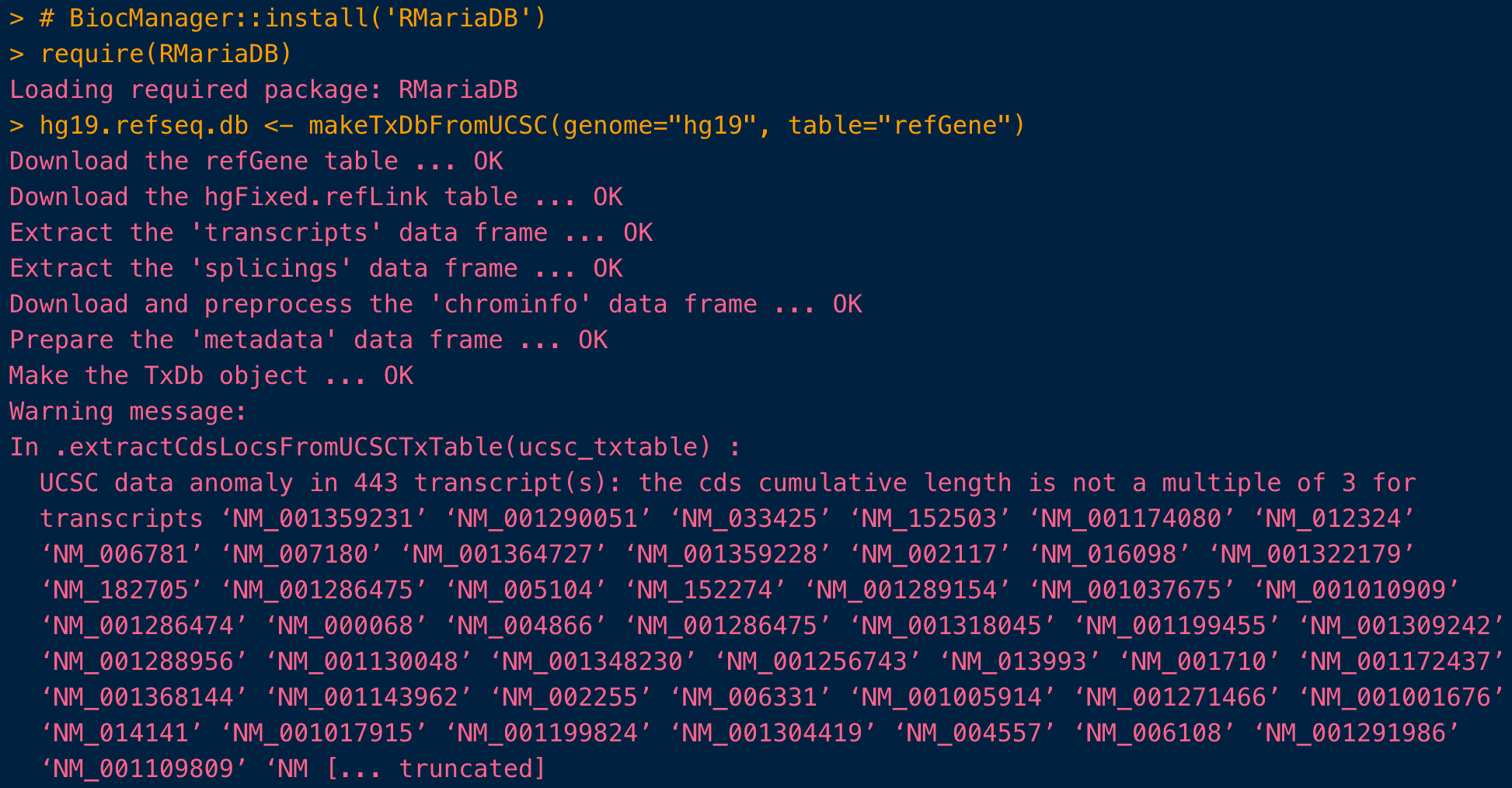
require(GenomicFeatures)
# makeTxDbFromUCSC()函数依赖RMariaDB这个包
# BiocManager::install('RMariaDB')
hg19.refseq.db <- makeTxDbFromUCSC(genome="hg19", table="refGene")
# 可能会遇到一个报错:namespace ‘DBI’ 1.0.0 is already loaded, but >= 1.1.0 is required =>自己升级
# remove.packages("DBI", lib="~/Library/R/3.6/library")
# packageurl <- "https://cran.r-project.org/src/contrib/DBI_1.1.0.tar.gz"
# install.packages(packageurl, repos=NULL, type="source")
然后可以对比一下:
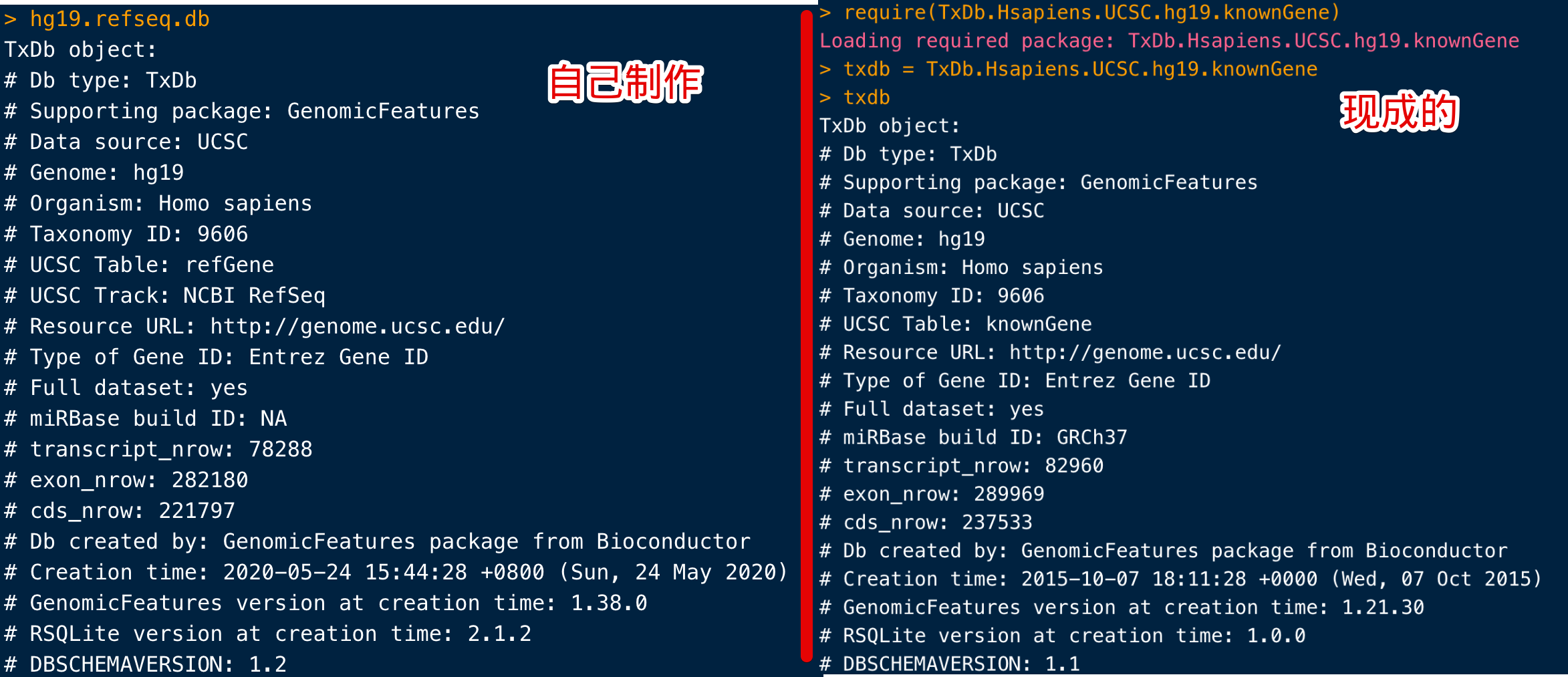
再比如自己下载GTF然后生成TxDb
以大豆(glycine_max)为例
# 下载
download.file('ftp://ftp.ensemblgenomes.org/pub/plants/release-47/gff3/glycine_max/Glycine_max.Glycine_max_v2.1.47.chr.gff3.gz',destfile = 'Glycine_max_v2.1.47.chr.gff3.gz')
# 解压
R.utils::gunzip('Glycine_max_v2.1.47.chr.gff3.gz')
# 制作
glycine <- makeTxDbFromGFF("Glycine_max_v2.1.47.chr.gff3")
有了TxDb怎么查看呢?
最详细的操作在官方文档:https://bioconductor.org/packages/release/bioc/vignettes/GenomicFeatures/inst/doc/GenomicFeatures.pdf
不管是从Bioconductor下载的还是自己制作的,都是一个GenomicFeatures对象
如果简单对名称操作,会返回这个注释文件的基本信息。要把TxDb当成一个数据库来对待,而不是一个简单的数据框或者矩阵。因此它的提取方法也会比较特别
- 如果想看其中包含的类目,可以用
columns(txdb) - 如果想指定提取转录本或外显子信息,可以:
transcripts(txdb) 或者 exons(txdb) - 如果想看全部的信息,可以:
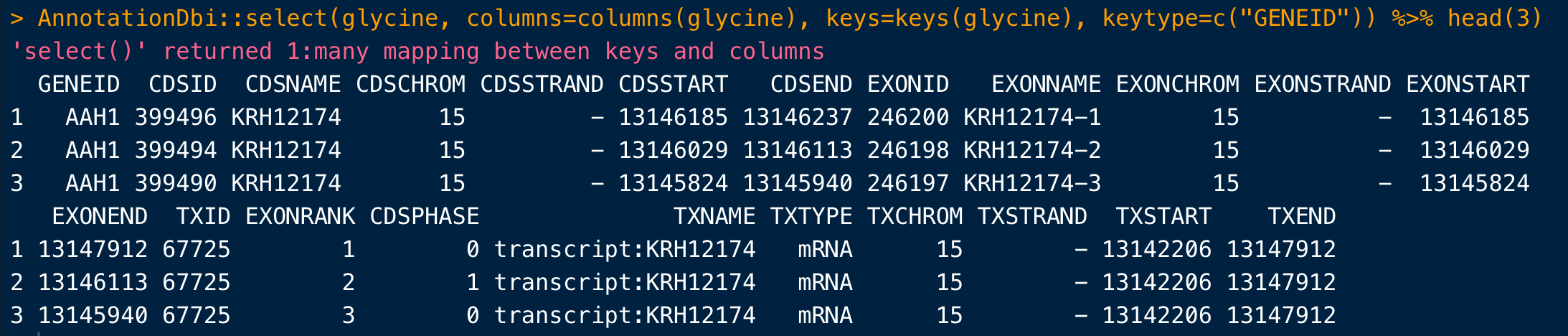
AnnotationDbi::select(glycine, columns=columns(glycine), keys=keys(glycine), keytype=c("GENEID"))
需要注意,如果使用这个select的时候,同时加载了tidyverse,那么同名的select就会发生冲突导致报错,这时可以用显式指定的形式来规范(如下图)
可视化
peak在整个染色体的分布
见:第3部分=》 使用covplot可视化BED数据
peak在某个窗口的结合谱图
一般有两种方式:一是直接使用BED文件,二是一步步手动进行
第一种:直接使用BED文件
peakHeatmap(f, weightCol="V5", TxDb=txdb,
upstream=3000, downstream=3000,
color=rainbow(length(f)))
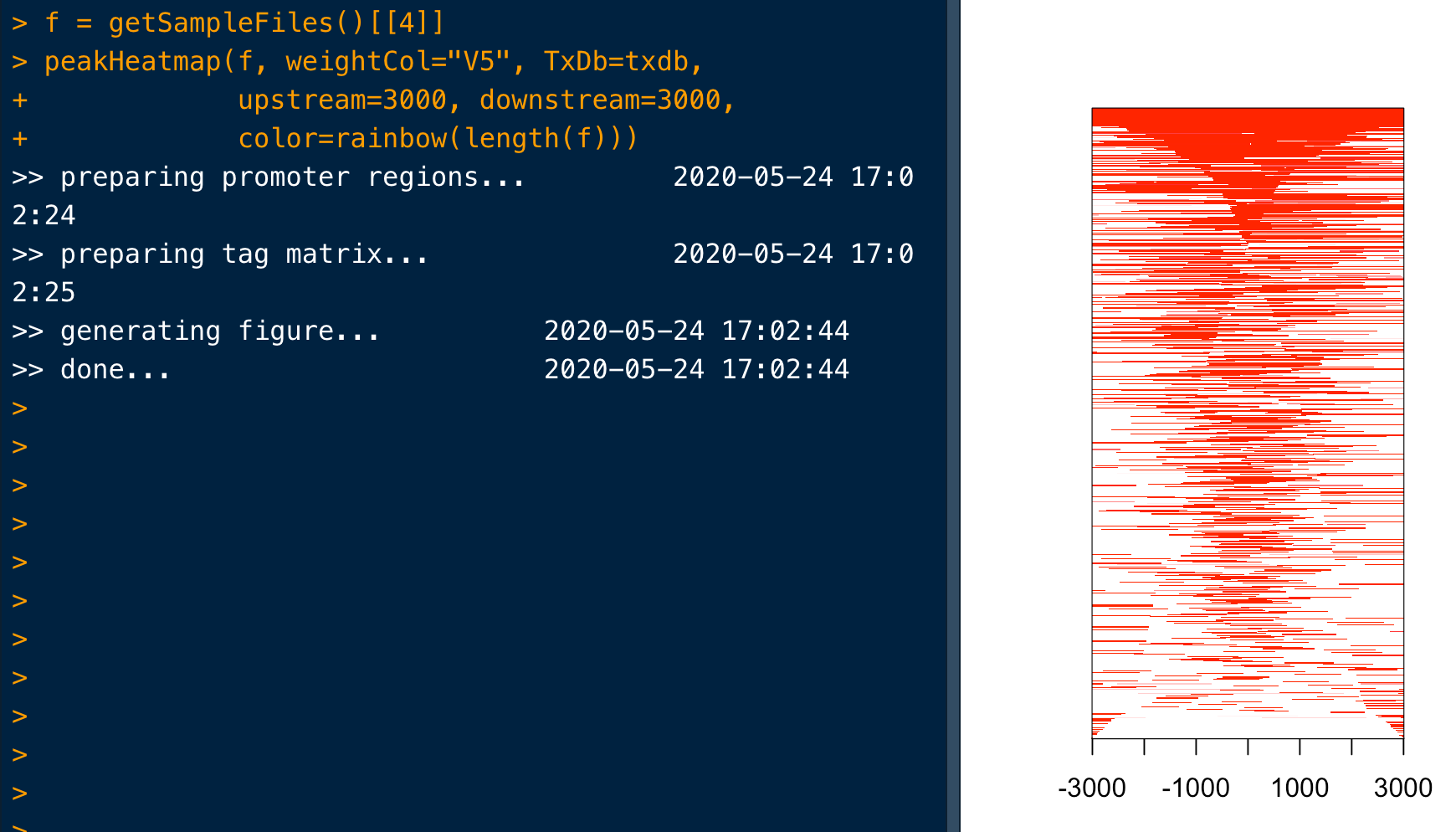
其实看运行日志也能看出来做了什么,首先根据转录起始位点指定上下游(也就是热图的窗口区间范围),然后把peaks比对到这个窗口,并生成矩阵以进行可视化
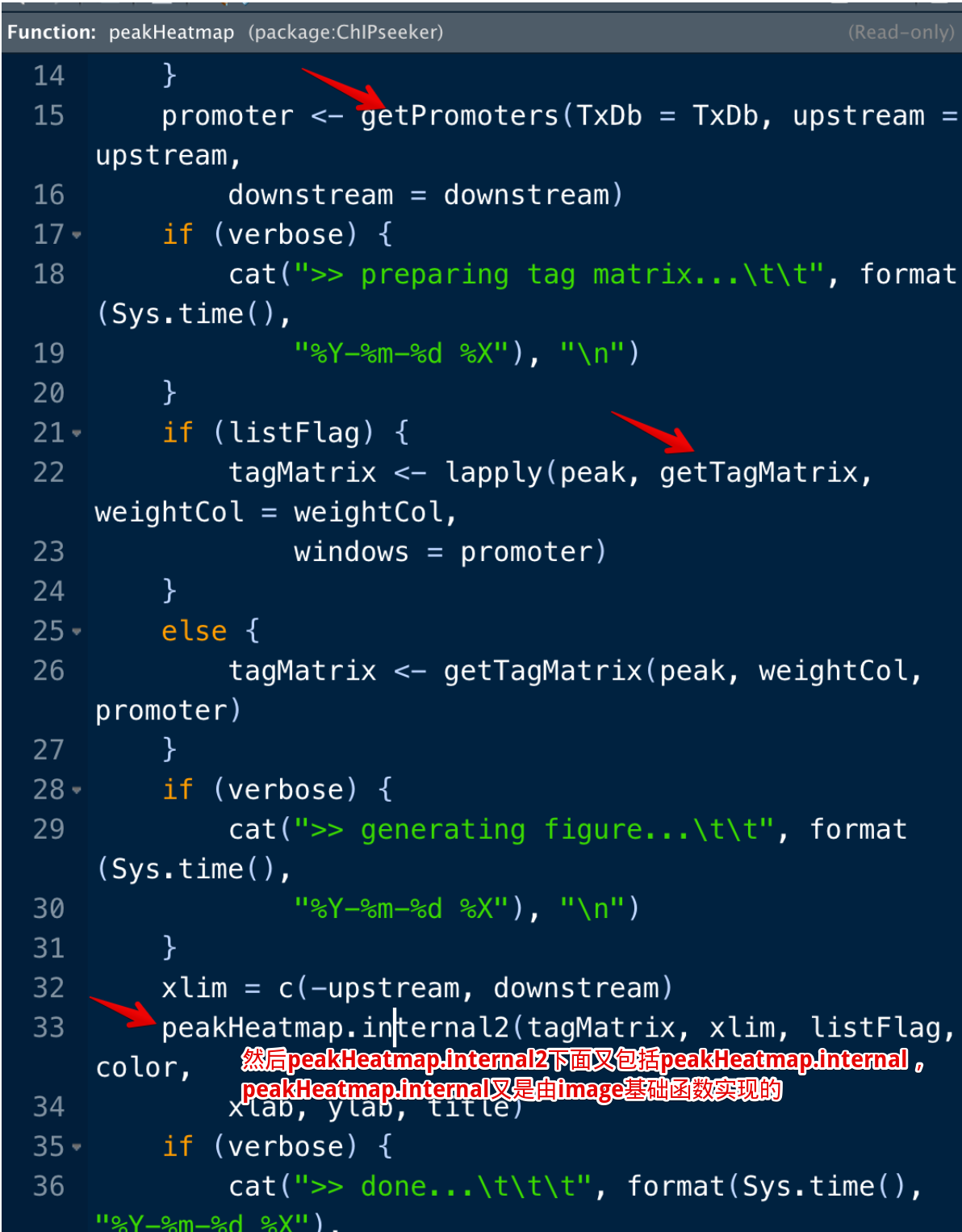
稍微查看一下这个peakHeatmap函数,就会发现以上说的几步:
当然,如果是多个文件也是可以的
files=getSampleFiles()
peakHeatmap(files, TxDb=txdb,
upstream=3000, downstream=3000,
color=rainbow(length(files)))
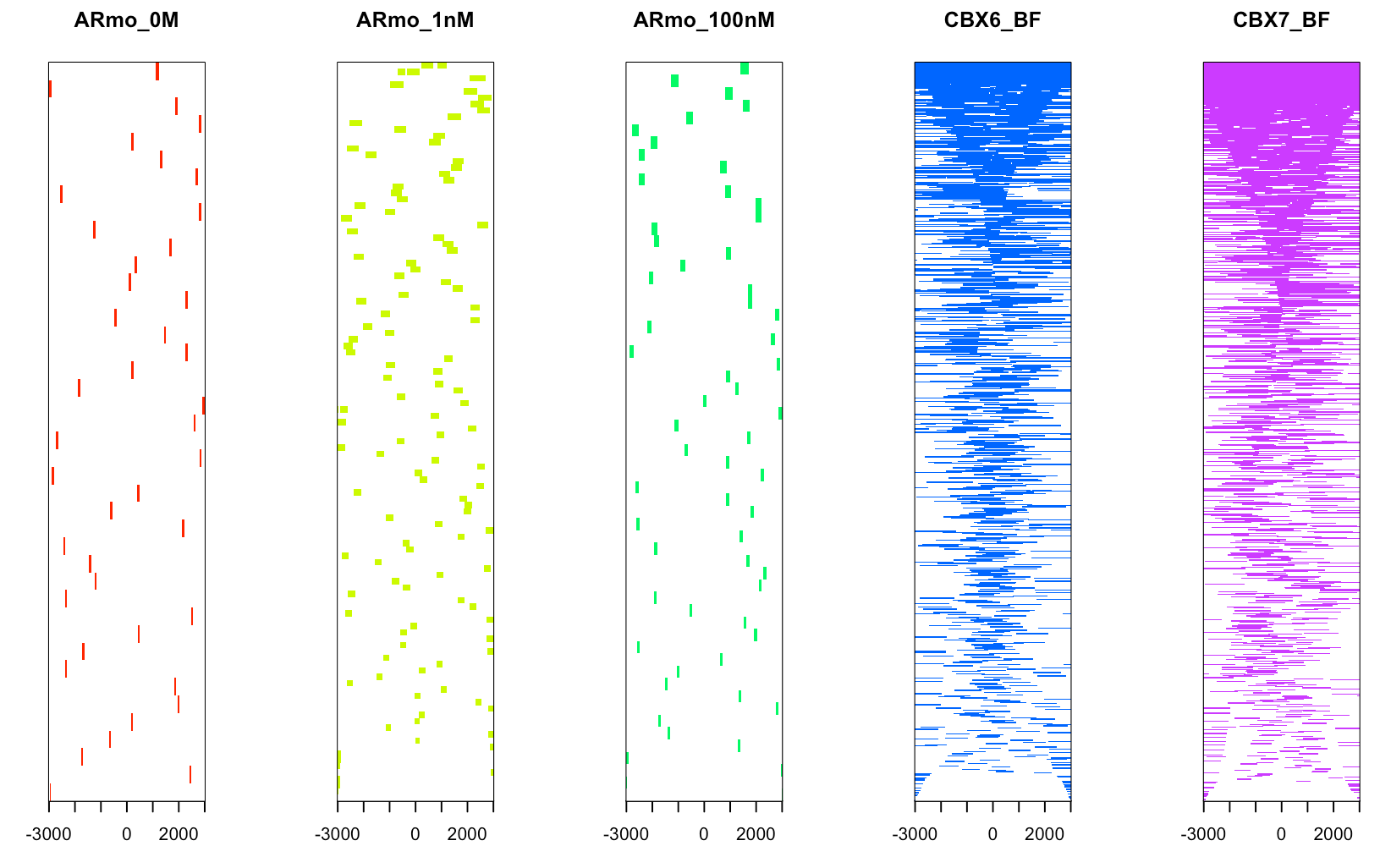
第二种:一步步手动进行
如果说第一种提供了一个打包好的计算过程,那么第二种就是把第一种拆分运行
promoter <- getPromoters(TxDb=txdb,
upstream=3000, downstream=3000)
tagMatrix <- getTagMatrix(f,
windows=promoter)
tagHeatmap(tagMatrix, xlim=c(-3000, 3000),
color="red")
看看结合的强度
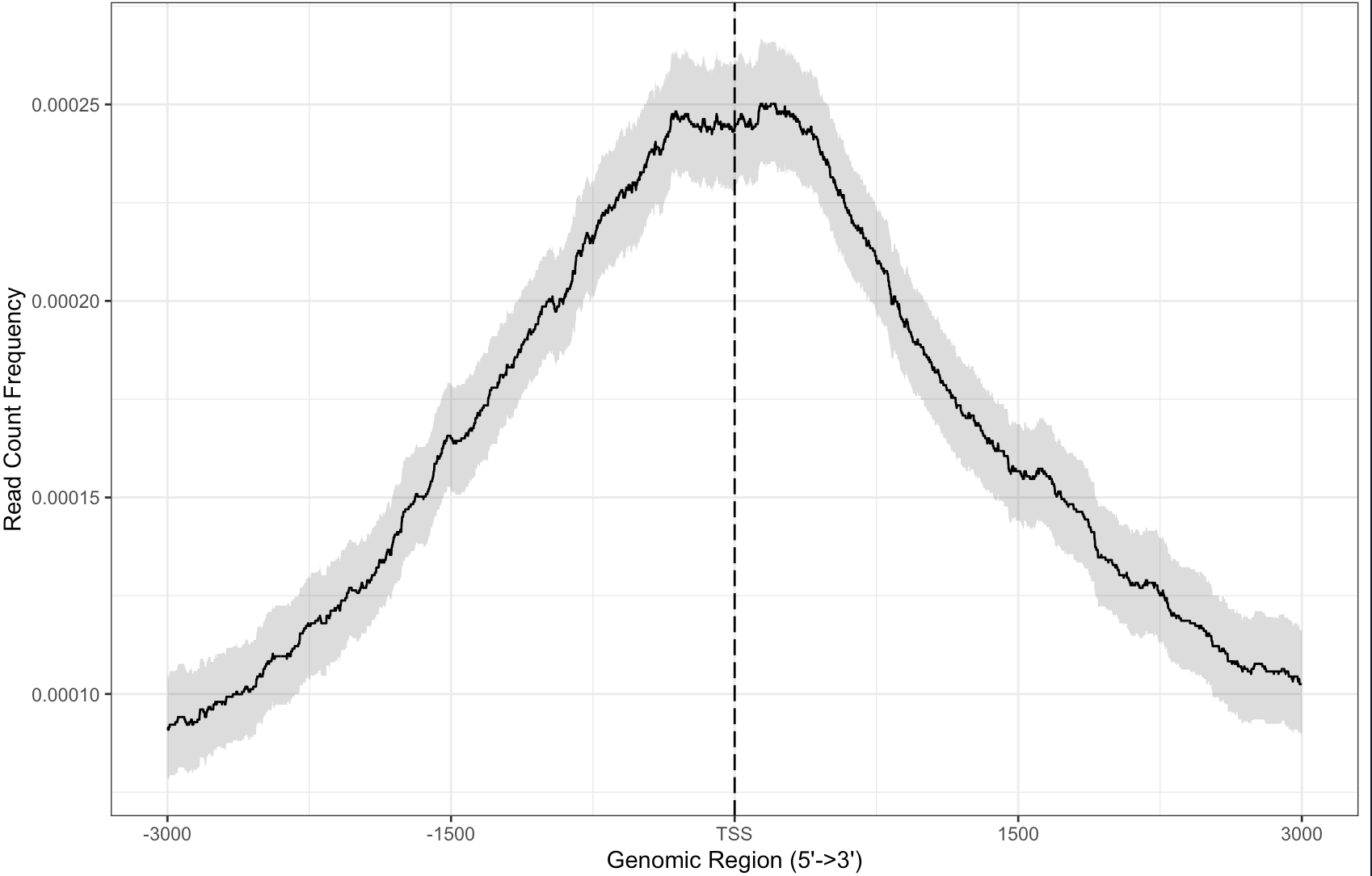
第一种:直接使用BED文件
plotAvgProf2(files[[4]], TxDb=txdb,
upstream=3000, downstream=3000,
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency",
conf = 0.95, resample = 1000)# 添加置信区间
第二种:手动进行
使用上面的tagMatrix计算结果
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency")
支持多个数据比较
tagMatrixList <- lapply(files, getTagMatrix,
windows=promoter)
# 添加置信区间并分面
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000),
conf=0.95,resample=500, facet="row")
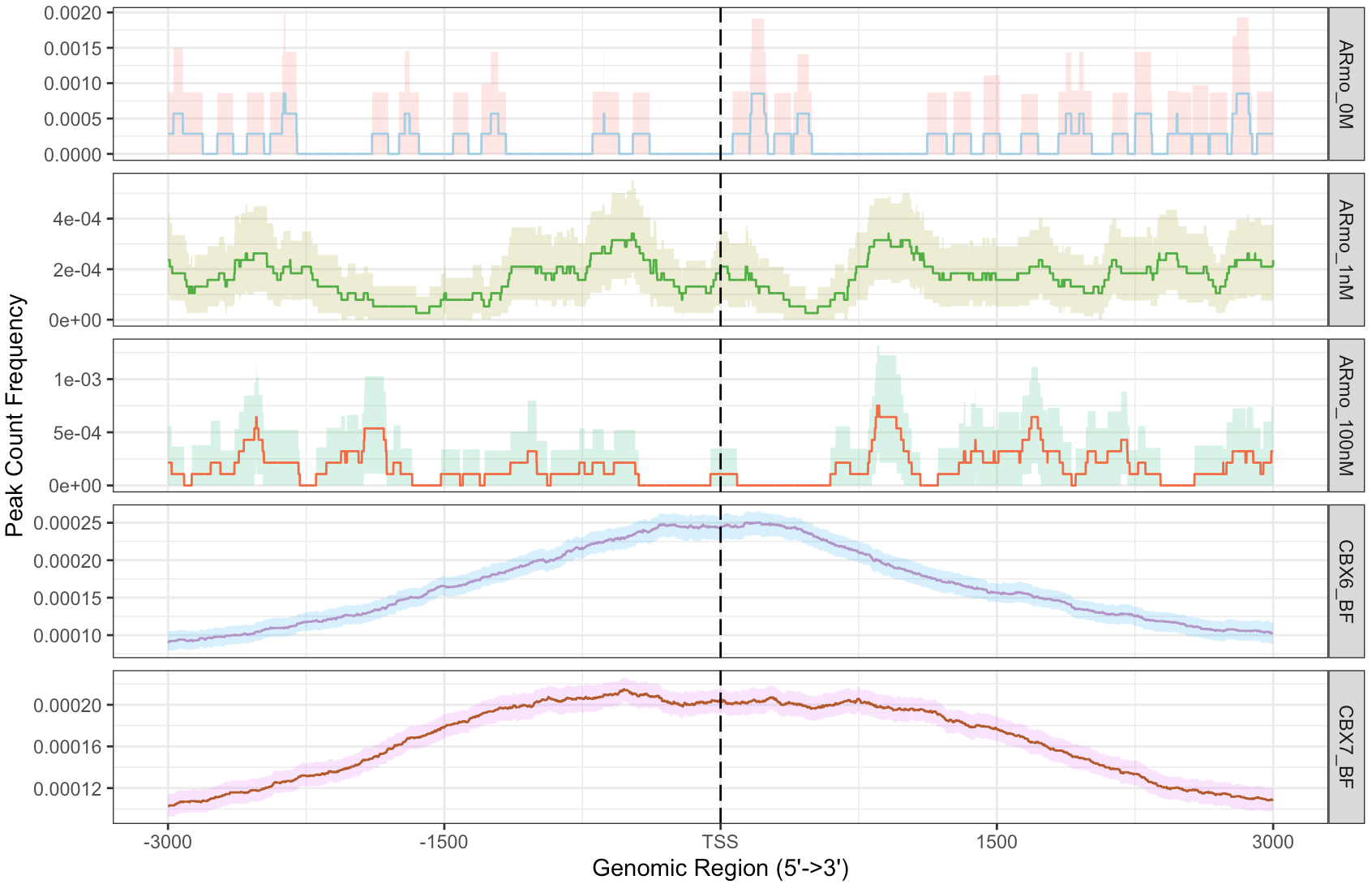
这个结果和上面peakHeatmap的结果一致,前3个样本不是调控转录的
除了关注转录起始位点(研究转录调控),还能看蛋白与外显子/内含子起始位置的结合谱,使用getBioRegion函数,可以指定'gene', 'transcript', 'exon', 'intron'
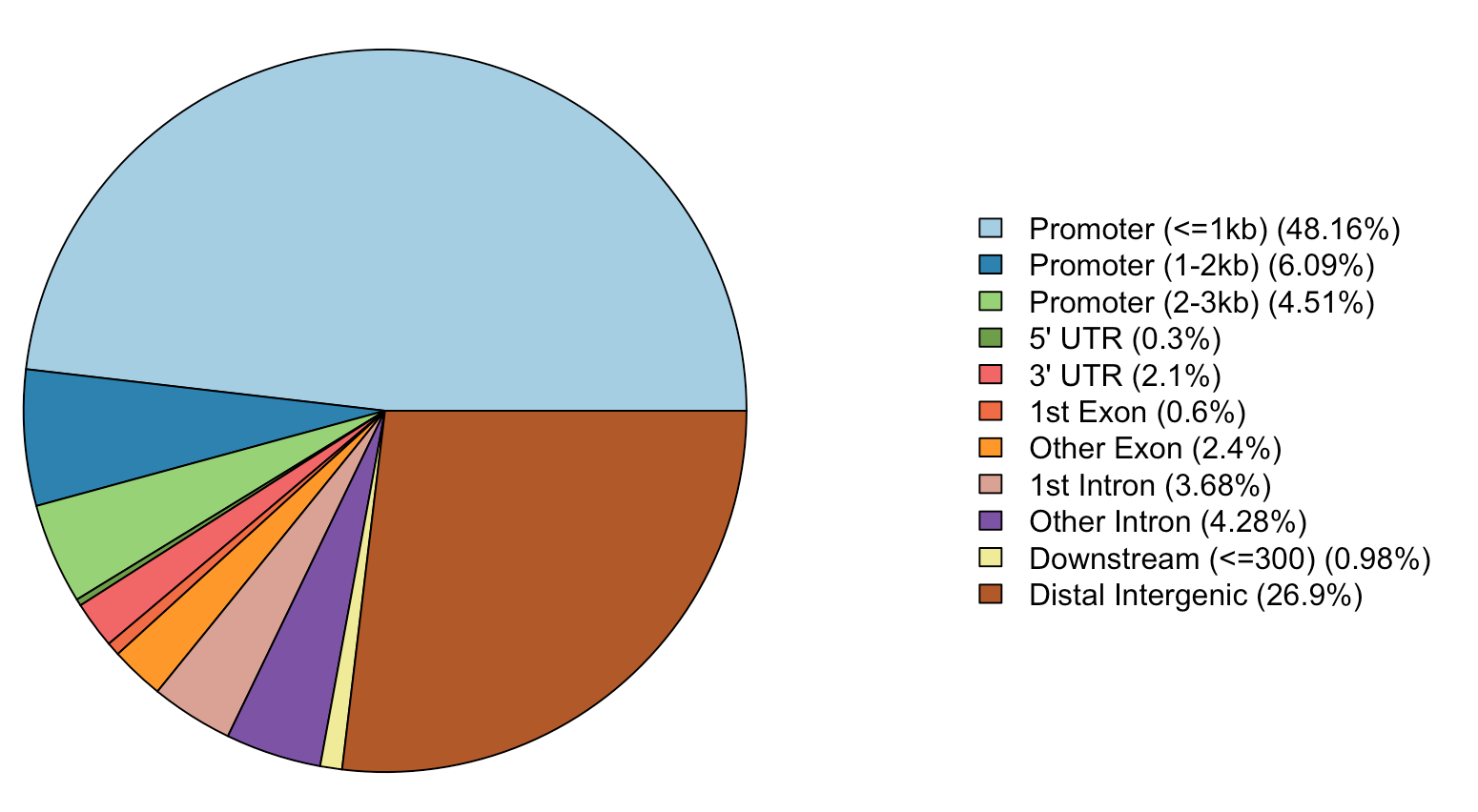
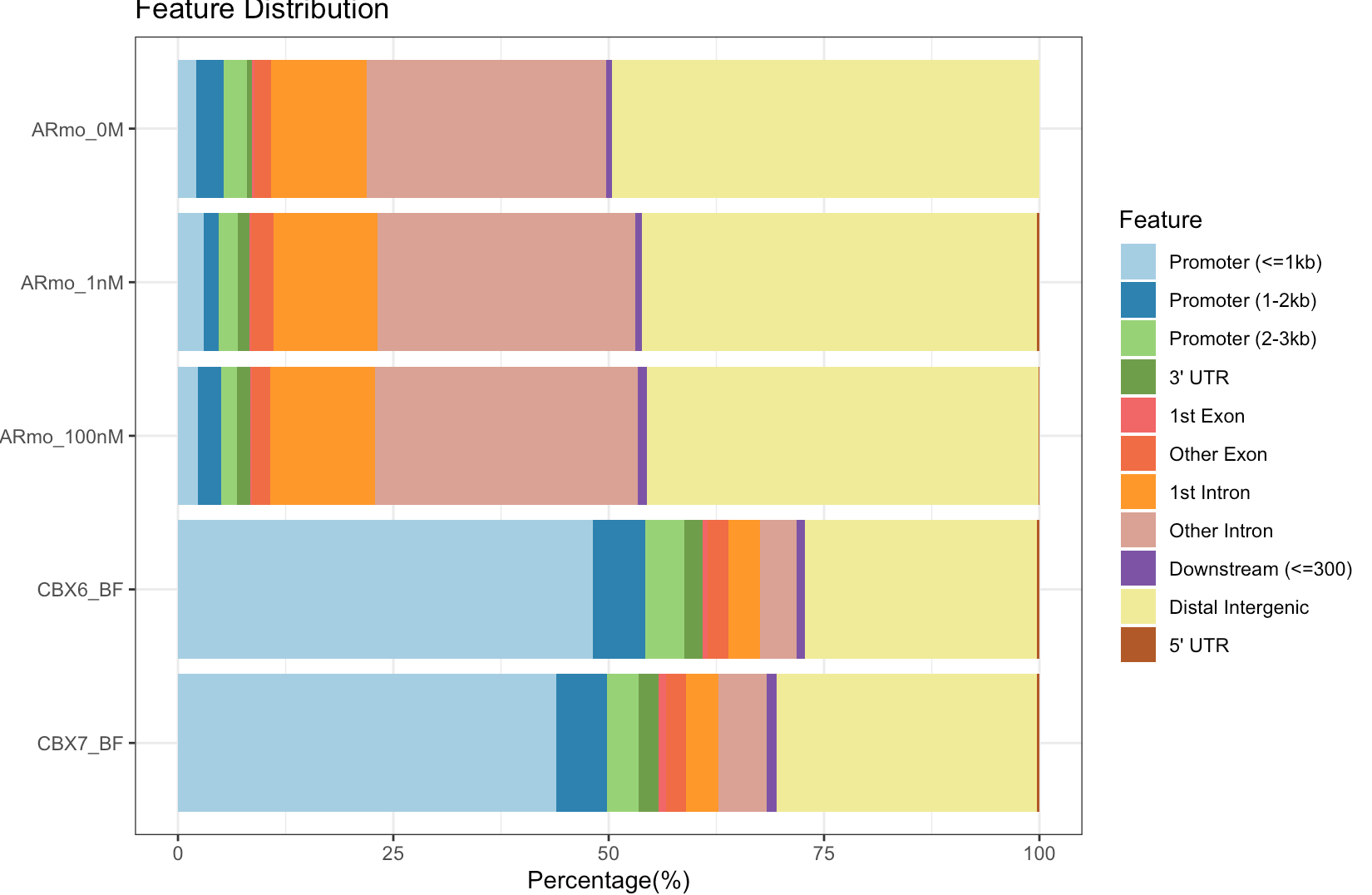
注释结果之注释类型一:genomic annotation
指peak在基因组的位置:落在什么地方,例如外显子、内含子或是5’ /3‘UTR
饼图
peakAnno <- annotatePeak(files[[4]],
tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Hs.eg.db")
plotAnnoPie(peakAnno)
柱状图
plotAnnoBar(peakAnno)
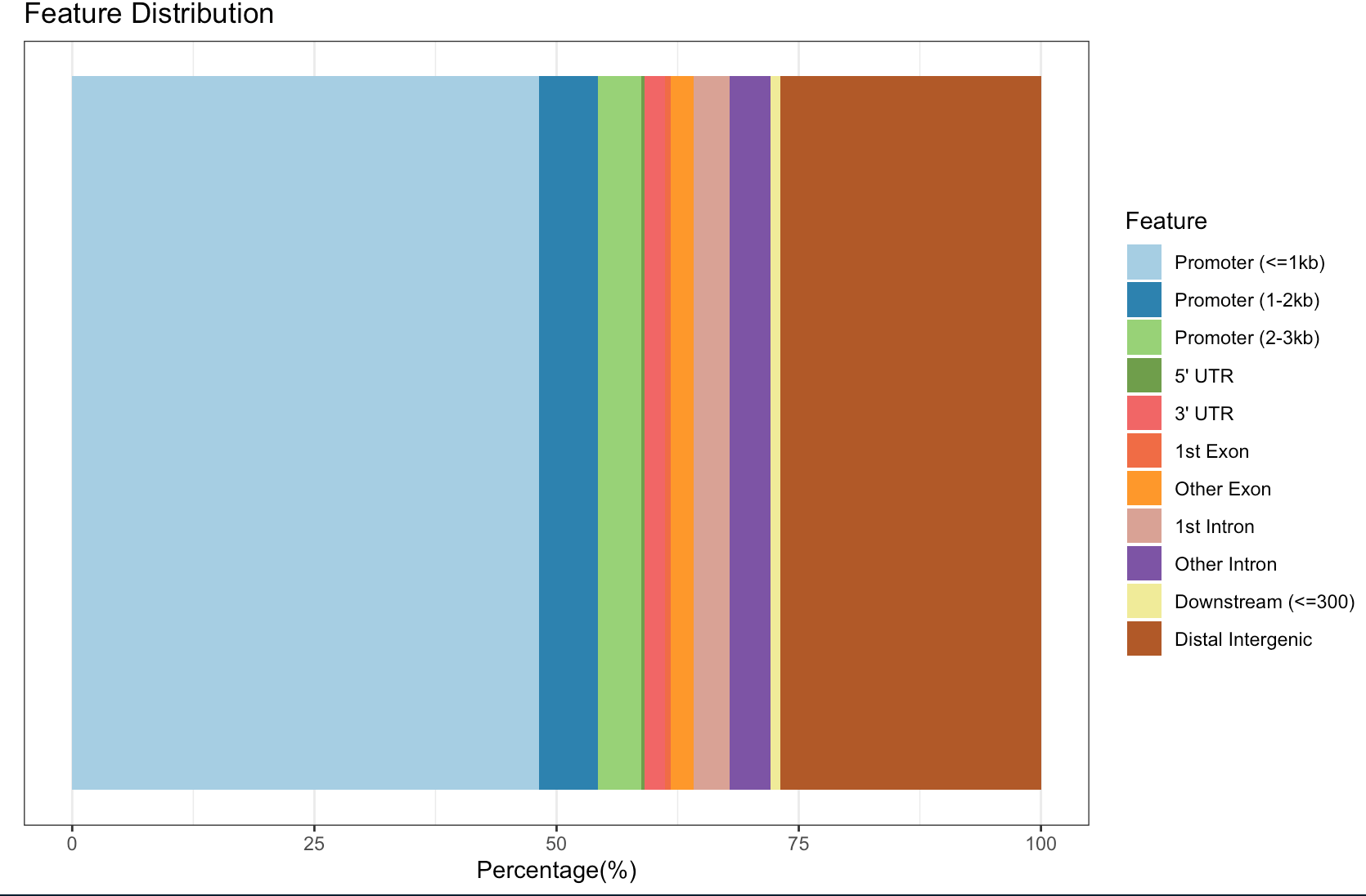
注意第一个问题:关于上图中的各种Features分类
看到这里的分类有下游(Downstream)但没有上游,这是因为Promoter定义为了转录起始位点(TSS)的上下游区域,包含了上游;另外这个下游是是基因间区的一部分,更确切是指紧接着基因的下游;这里的上游和下游其实都是基因间区,单独拿出来是因为和基因直接连接,是很近的区域=》近端基因间区
当然,基因间区还包含更远的间区(Distal intergenic)=》远端基因间区
默认下游的范围是3kb,但是可以自己调整
# 比如调成500
options(ChIPseeker.downstreamDistance = 500)
还有一个需求就是:自定义分类
# 依然是设置options,用于总结结果
f2=getSampleFiles()[[5]]
options(ChIPseeker.ignore_1st_exon = T)
options(ChIPseeker.ignore_1st_intron = T)
options(ChIPseeker.ignore_downstream = T)
options(ChIPseeker.ignore_promoter_subcategory = T)
x=annotatePeak(f2)
plotAnnoPie(x)
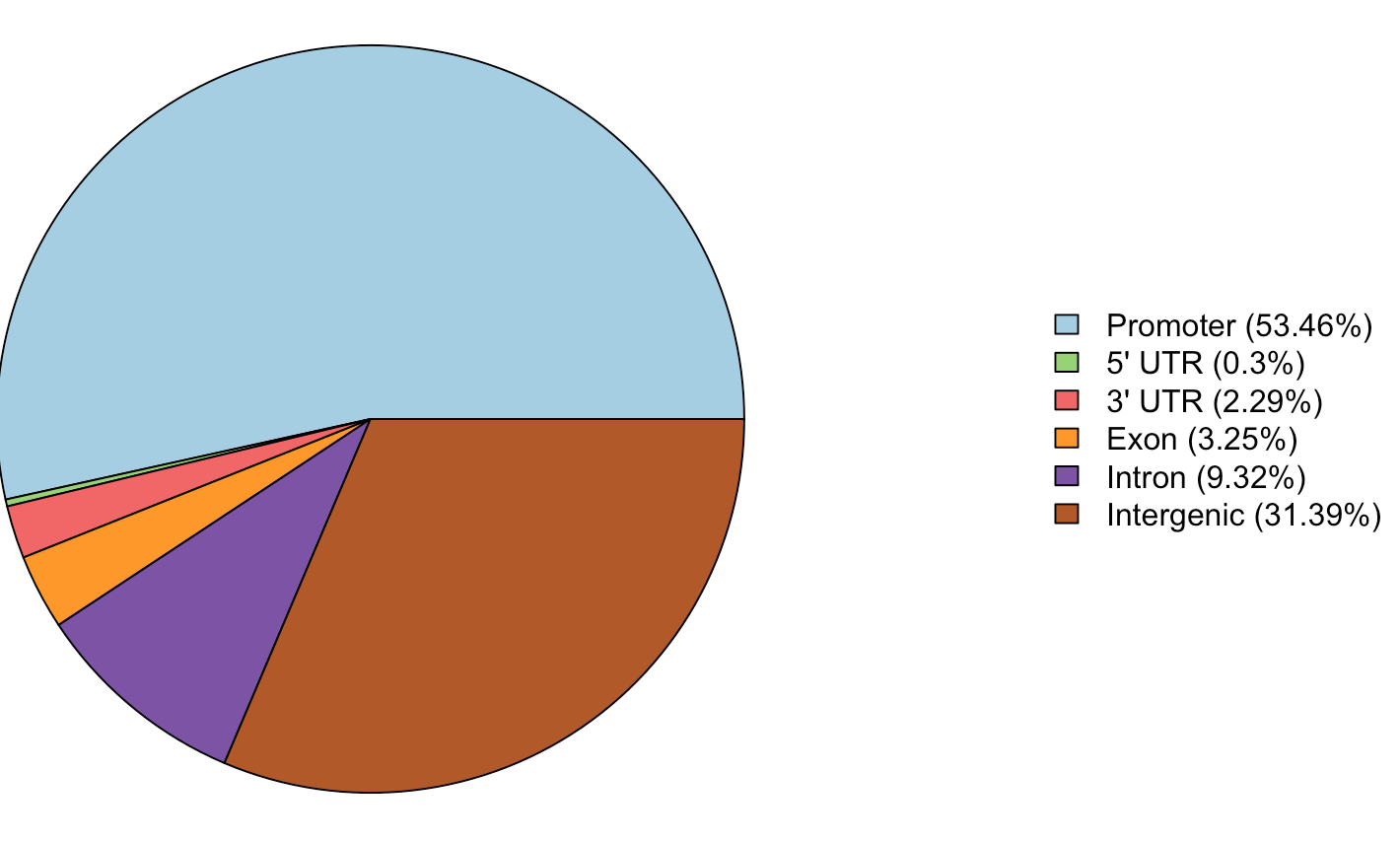
注意第二个问题:peak的位置可能不是唯一的
这是因为,一个peak所在的位置,可能是一个基因的外显子,同时又是另一个基因的内含子。为了解决这个问题,有以下几种方案:
- 第一种:使用参数
genomicAnnotationPriority指定优先顺序
默认顺序是:Promoter => 5’ UTR => 3’ UTR => Exon => Intron => Downstream => Distal Intergenic
- 第二种:饼图+韦恩图
vennpie(peakAnno)
优点是:直观;缺点是:无法显示全部的信息
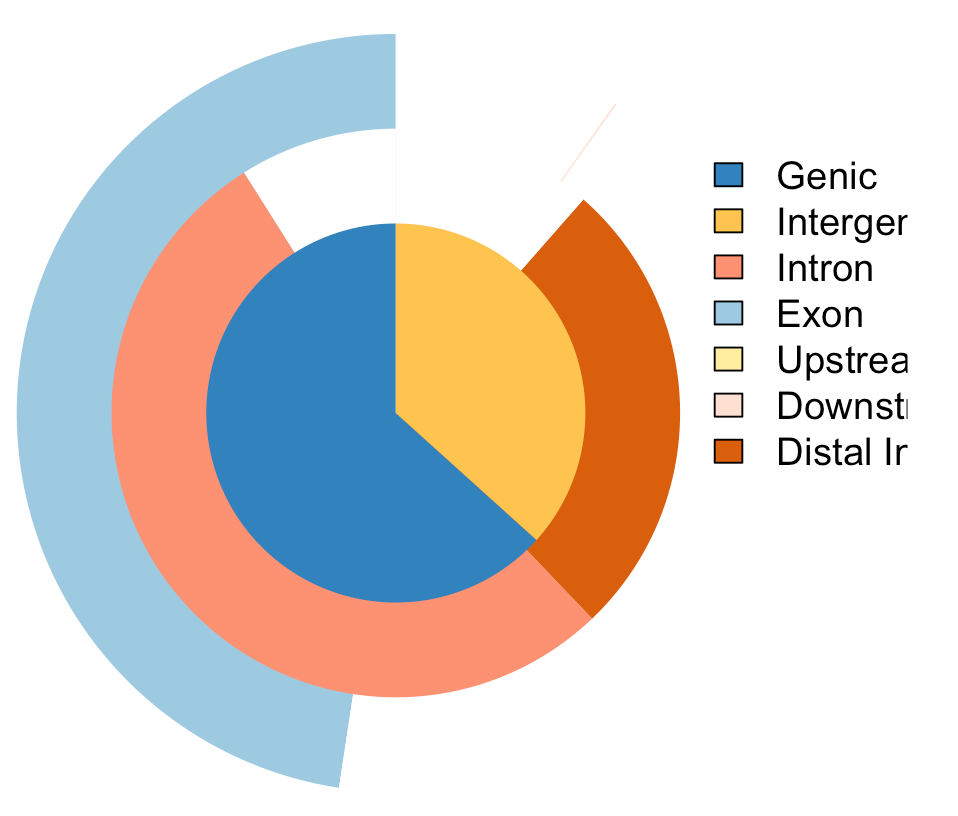
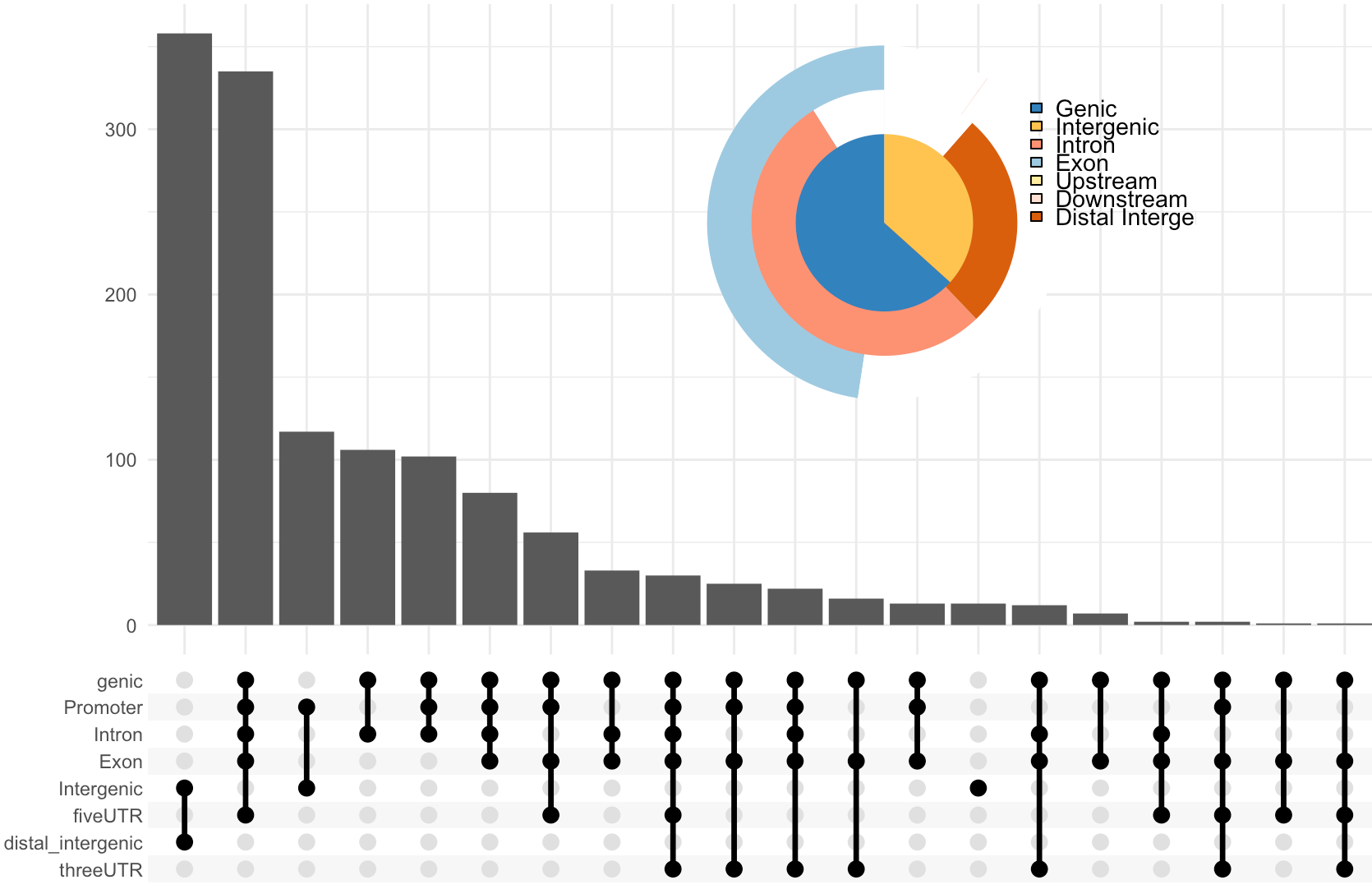
- 第三种:UpSetR + vennpie
upsetplot(peakAnno, vennpie=TRUE)
多个文件的区域注释
peakAnnoList <- lapply(files, annotatePeak,
TxDb=txdb,tssRegion=c(-3000, 3000))
plotAnnoBar(peakAnnoList)
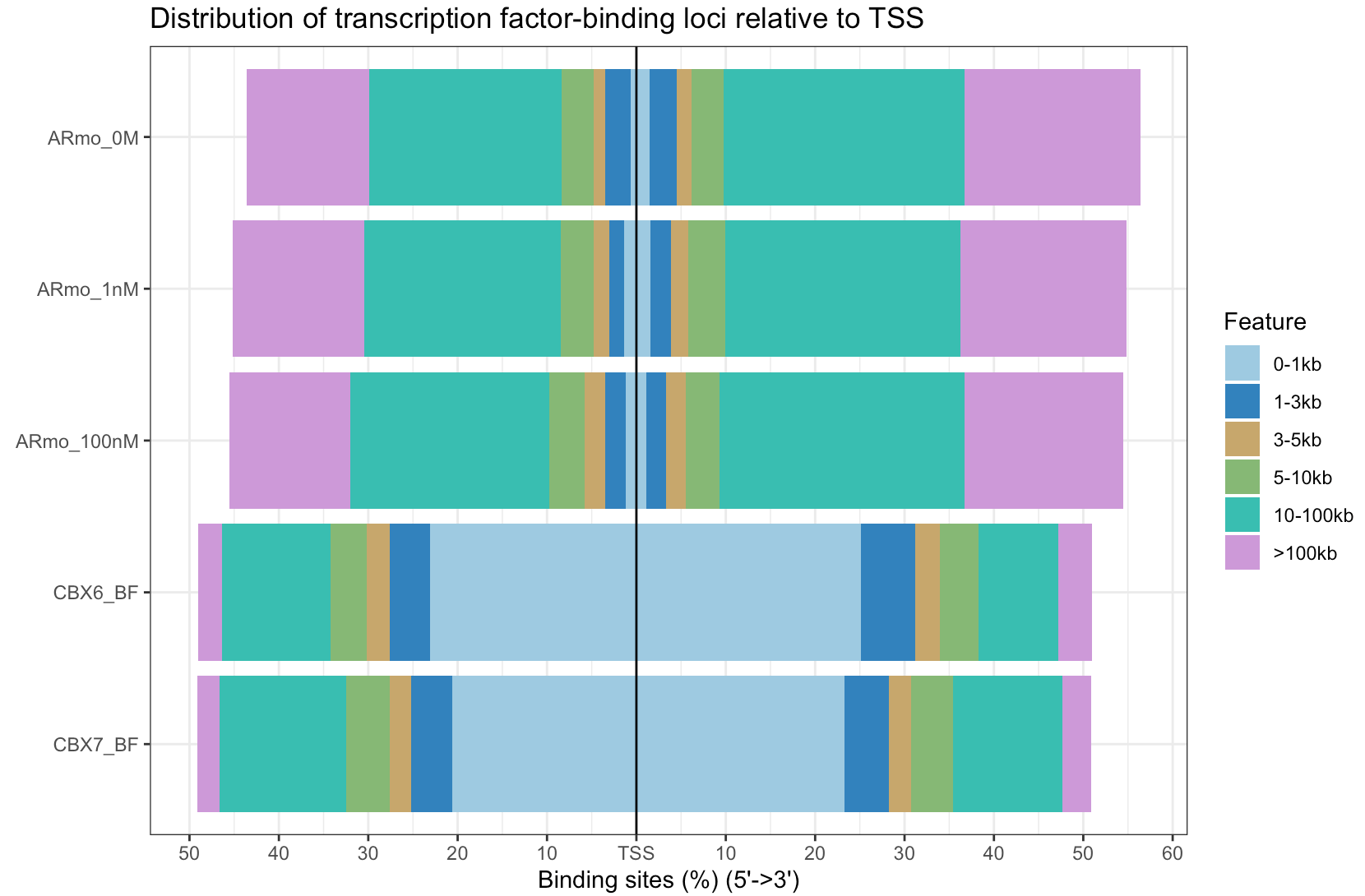
注释结果之注释类型二:nearest gene annotation
指peak最近的基因:不管peak落在内含子、基因间区还是其他位置,按照peak相对于转录起始位点的距离,都能找到一个离它最近的基因
plotDistToTSS(peakAnno,
title="Distribution of transcription factor-binding loci\nrelative to TSS")
同样也支持多个文件
plotDistToTSS(peakAnnoList)
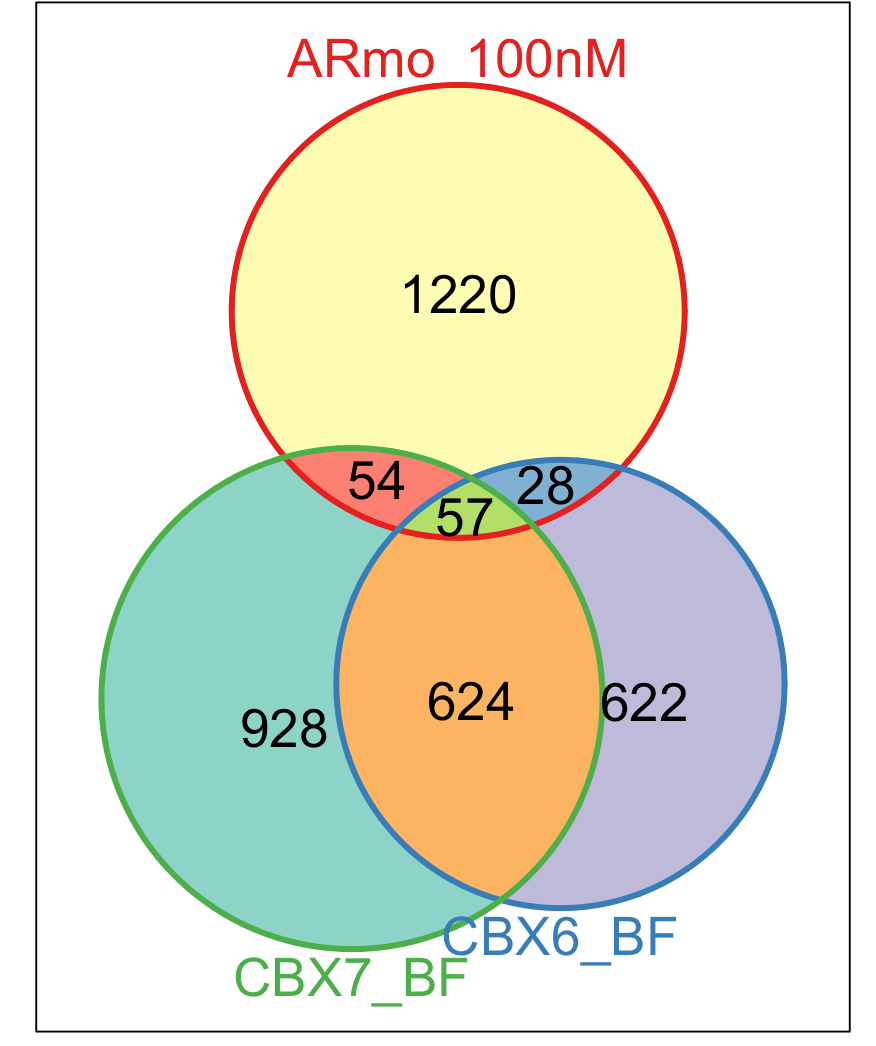
距离最近的基因在不同样本的交集
# 先得到基因列表
genes <- lapply(peakAnnoList, function(i)
as.data.frame(i)$geneId)
> names(genes)
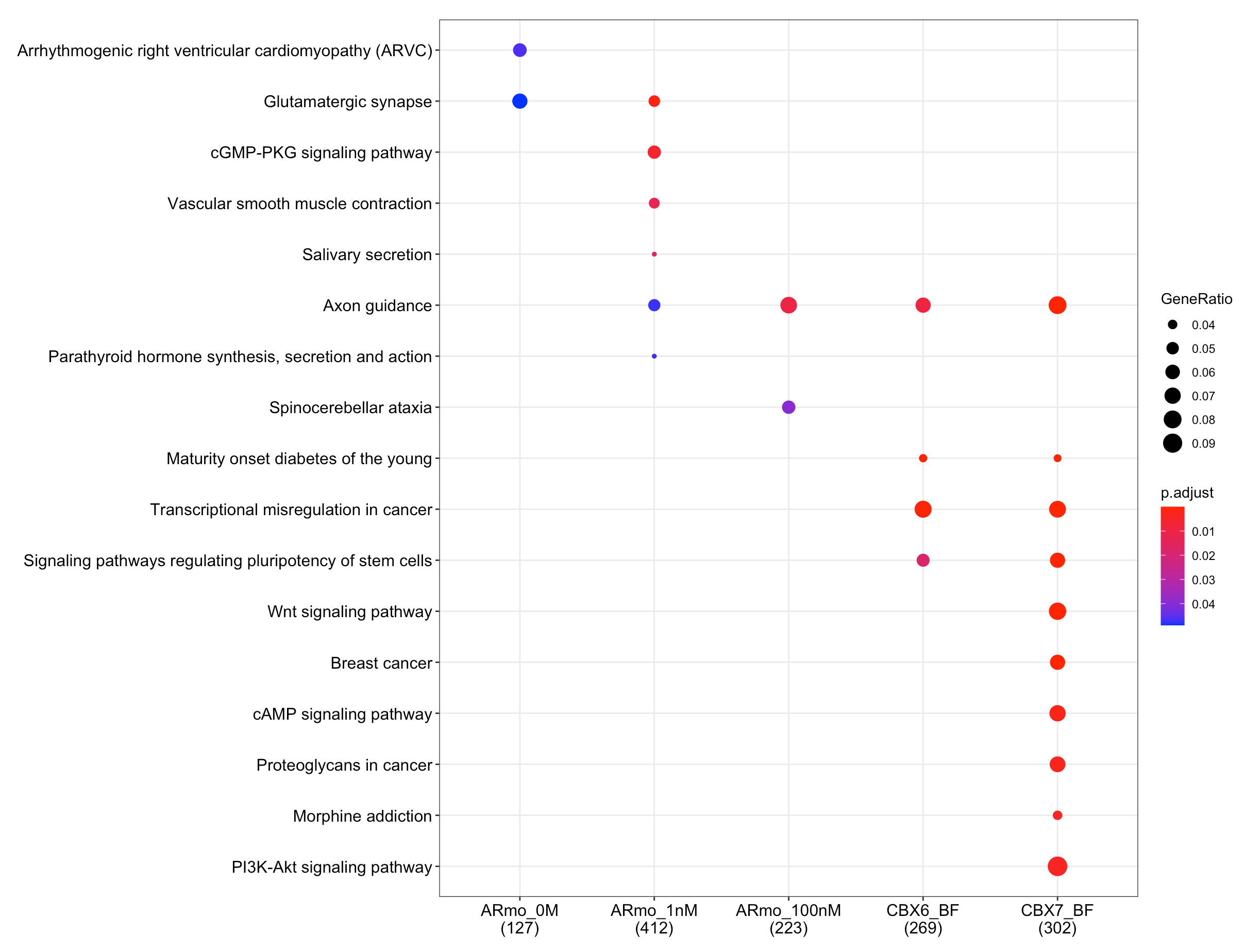
[1] "ARmo_0M" "ARmo_1nM" "ARmo_100nM" "CBX6_BF" "CBX7_BF"
# 然后作图(需要借助Vennerable包)
devtools::install_github("js229/Vennerable")
library(Vennerable)
vennplot(genes[2:4], by='Vennerable')
基因注释 + 富集分析
利用ChIPseeker的seq2gene 将peak的位置与所有的基因关联起来【包括 host gene (exon/intron), promoter region and flanking gene from intergenomic region】,然后用clusterProfiler拿这些基因跑ORA,做富集
require(clusterProfiler)
bedfile=getSampleFiles()
# 将bed文件读入(readPeakFile是利用read.delim读取,然后转为GRanges对象)
seq=lapply(bedfile, readPeakFile)
genes=lapply(seq, function(i)
seq2gene(i, c(-1000, 3000), 3000, TxDb=txdb))
cc = compareCluster(geneClusters = genes,
fun="enrichKEGG", organism="hsa")
dotplot(cc, showCategory=10)